2. Materials and methods
2.1 Atmospheric chemistry–transport model (ACTM)
Atmospheric CH4 concentrations were simulated using the atmospheric general circulation model (MIROC-AGCM, version 4.0) at a horizontal resolution of approximately 2.8125° × 2.8125° (a triangular truncation of 42 horizontal wavenumbers: T42) with 67 hybrid vertical layers between the Earth's surface and 0.0128 hPa (∼ 80 km) for 1988–2017 after a spinup for 6 year (1982–1987). The initial CH4 distribution on 01 January 1982 was taken from an earlier simulation of 29 year. More detail about MIROC4-ACTM can be found in the literature (Watanabe et al. 2011; Patra et al. 2018). The following continuity equation is solved for the time evolution of CH4 burden (BACTM) at each grid and altitude in the Earth's atmosphere.
where
-
S0 = emissions of CH4 obtained from bottom-up emission inventories and terrestrial ecosystem model simulations (a priori).
-
L = loss of CH4 by reaction with hydroxyl radicals (OH), atomic chlorine (Cl), and oxygen in an excited state (O(1D))
-
∇φ = transport of CH4 by advection, convection, and diffusion
, and
t represent index for longitude, latitude, altitude, and time. Global mean
BACTM closely represents the simulated atmospheric concentration
DACTM (
Table 1).

The MIROC4-ACTM simulated horizontal winds (U and V) and temperature (T) are nudged to the Japan Meteorological Agency reanalysis fields (Kobayashi et al. 2015) at 2–61 vertical levels for better representation of synoptic-scale transport features. MIROC4-ACTM is extensively validated for large-scale interhemispheric transport using sulfur hexafluoride (SF6) in the troposphere (supplementary Fig. S1) and the Brewer–Dobson circulation in the stratosphere using SF6 and CO2 derived age of air (Patra et al. 2018). The prescribed three-dimensional OH field from Spivakovsky et al. (2000) is used for calculating the chemical loss of CH4 in the atmosphere. The global mean OH is optimized by applying a single scaling factor of 0.92 to the monthly varying OH field to simulate the observed CH3CCl3 decay rate in the troposphere (Fig. S2). The northern-to-southern hemispheric OH ratio is close to 1 (Patra et al. 2014). This case is considered as the “control case”. Stratospheric CH4 losses due to reaction with OH, O(1D), and Cl radicals are calculated using their concentration fields simulated by ACTM's stratospheric model run (Takigawa et al. 1999). The CH4 loss by reaction with Cl in the troposphere is not included due to the lack of sufficient information on the abundance of Cl (Hossaini et al. 2016; Gromov et al. 2018; Nisbet et al. 2019).
2.2 A priori CH4 emissions
The a priori CH4 emissions are prepared monthly by combining the emissions from all anthropogenic and natural sectors and by subtracting the surface sinks due to bacterial consumption in the soil (Fig. 1; Table S1). The anthropogenic emissions for major sectors (fugitive, enteric fermentation and manure management, solid waste, and wastewater handling) are used from the Emission Database for Global Atmospheric Research, version 4.3.2 inventory (EDGARv4.3.2) (Janssens-Maenhout et al. 2019). The EDGARv4.3.2 inventory provides emissions for individual sectors at the spatial resolution of 0.1° × 0.1° on an annual basis for 1970–2012 and on a monthly basis for 2010 only. We applied the seasonality of individual sectors from 2010 to the yearly emissions of the respective sectors during 1980–2012. The growth rate for 2011–2012 has been used to interpolate the EDGARv4.3.2 inventory emissions from 2013 to 2017. Monthly emissions for biomass burning are taken from the Global Fire Database (GFEDv4s) (van der Werf et al. 2017) and Goddard Institute for Space Studies emissions (Fung et al. 1991) (scaled by ×0.315). A climatological mean (1997–2016: 15.6 Tg yr−1) is used for the period from 1988 to 1996 when the GFEDv4s dataset is unavailable.

A process-based model of the terrestrial biogeochemical cycle, Vegetation Integrated Simulator of Trace gases (VISIT) (Ito 2019; Ito et al. 2019) is used to estimate the wetland and rice emissions on monthly basis using two different schemes developed by Walter and Heimann (2000) (referred to as “WH” scheme in the text) and Cao et al. (1996) (referred to as “Cao” in the text). The detailed simulation protocols are discussed in Ito (2019). However, we understand the need for more accurate wetlands area mapping, e.g., over the tropical (Amazonia, Pantanal, Nile, Congo, Zambia, Indonesia, and Papua New Guinea) as well as the mid-latitudes and boreal regions (Nisbet et al. 2020). The other emissions (e.g., ocean, termites, and mud volcano) are taken from the TransCom–CH4 intercomparison experiment (Patra et al. 2011). Figure 1 shows the spatial distribution of the total a priori emissions and regional total time series of selected aggregated sectors (please note the six major sectorial aggregations in the legend). Table S1 summarizes the global total sectorial emissions during different decades.
2.3 Atmospheric CH4 data from surface sites, aircraft flights, and remote sensing satellite
We use the surface observations of CH4 dry air mole fraction from the National Oceanic and Atmospheric Administration (NOAA) cooperative global air sampling network sites (Dlugokencky 2019; Dlugokencky et al. 2020) for estimating monthly mean emissions by inversion. Figure 2 shows the site locations. These measurement sites are chosen so that the temporal data gap is minimized for the period of inverse calculation, 1988–2016. ACTM simulated results are sampled at the nearest model grid from the output at hourly average values. For the observed and simulated time series at each station, digital filtering was applied for the monthly mean values using a cut-off length of 24 months and six harmonics (Nakazawa et al. 1997). The data gap has been filled by using the fitting curve. This gap filing approach was required as there are data gaps of 1–2 months at many sites during the long operation period (1988–2016).

The conventional approach for validating a posteriori emission is to derive a robust metric using a set of independent measurements (not used in the inversion). A long-term ongoing aircraft measurement program over Japan since 1988 by Tohoku University (Umezawa et al. 2014) gives an excellent opportunity to validate the emission trend estimated by our inverse modeling. For this analysis, we have combined observations from two sub-programs of Tohoku University: 1) measurements below 4 km are conducted over adjacent areas of Sendai airport (38.1°N, 140.9°E), and 2) measurements above 4 km are made along commercial airliner flights between Sendai and Fukuoka (33.6°N, 130.4°E) and between Sendai and Sapporo (42.8°N, 141.7°E). Only a part (region: 36–42.8°N, 136–141.7°E) of the whole data from the commercial aircraft observation are used in the analysis. The aircraft data are available on the Tohoku University CH4 scale (TU1987 scale), which is converted into the World Meteorological Organization scale by subtracting 0.5 ppb (Fujita et al. 2018; Aoki et al. 1992). It is emphasized that the observation location is downstream of continental East Asia in terms of atmospheric transport, thereby the dataset being advantageous in addressing emission changes in the region.
Total column dry air mole fractions of CH4 (XCH4) were observed using the Thermal And Near-infrared Sensor for carbon Observation (TANSO) instrument, comprising a Fourier transform spectrometer, onboard the Greenhouse Gases Observing Satellite (GOSAT), launched by Japan Aerospace Agency in 2009 (Kuze et al. 2016). We have used here the full physics retrievals of the National Institute for Environmental Studies version 2.72 (NIESv2.72) (Yoshida et al. 2013). Vertical profiles from MIROC4-ACTM simulations are sampled at the observation location and time and convolved with the retrieval of a priori vertical profile and column averaging kernels. The individual XCH4 retrievals and model simulations are aggregated in 2.5° × 2.5° grid boxes for presentation here.
2.4 Time-dependent inverse model for 53 land regions
Bayesian inversion is one of the powerful statistical tools to correct or refine our prior knowledge with the assimilation of data. We used a time-dependent Bayesian inverse model based on our earlier study (Patra et al. 2016), with a modification of a posteriori loss correction (Section 2.5). The inversion is conducted over the 53 partitioned land regions (Fig. 2) and estimated “total CH4 emissions” at a monthly time interval. Therefore, all interpretation about which source is changing comes from the priors. The CH4 emission is not optimized over the ocean region because of its small contribution (∼ 18 Tg yr−1) to the total global emissions. The source strengths are predicted by the least squares solution of the equation G.S = D by assuming the linear relations between emissions (S) and concentrations (D), where G is the Green's function that define the regional source–receptor relationship. The following equations are used to estimate the emissions and associated covariance (Cs):
where
S0 = regional prior sources,
CS0 = prior source covariance (square of uncertainty),
DObs = atmospheric observations,
DACTM = forward model simulations using a priori emissions, and
CD = data covariance.
The prior and data uncertainties indicate the relative weights in each set of information. The CS0 is set at 50 % of regional emissions. These flux regions are assumed to be uncorrelated to each other; hence, the off-diagonal elements of the CS0 matrix are set to 0. The inversion is also run at different CS0 (70 % of regional emission, constant at 30 and 60 Tg yr−1) to check the robustness of our set up. Very close agreement of a posteriori emissions for the three sensitivity simulations suggests that our inversion system is constrained well by atmospheric data than following the a priori assumptions. The CD is obtained by adding instrumental error (fixed at 5 ppb) to the transport model representation error, estimated as the monthly residual standard deviations (RSDs) of the daily mean time series. RSDs are calculated as the differences between the original time series and fitted time series that exclude the synoptic variability (Nakazawa et al. 1997). The model representation error arises due to the inability of the coarse ACTM resolution to simulate site-specific concentration gradients. Typical CD values are 5.3, 13.3, and 38 ppb at SYO, KEY, and TAP, respectively.
The sign of correction fluxes (S-S0) is decided by the differences between the observations and simulations using prior sources (the final term of Eq. 2). Although the atmospheric observations are not available over some regions, their fluxes are estimated as a combination of weighted basis function signals and mismatches between the atmospheric data and model simulations for the other regions (see Eq. 2). This helps to maintain the global mass balance between surface CH4 fluxes and the atmospheric CH4 burden.
The regional basis functions are calculated using the difference between a background CH4 tracer with seasonally varying emission (no interannual variations) and the background plus a unitary regional emission pulse over a month. The basis functions consist of the pattern of emissions (Fig. 1 map) within each of the inversion region (Fig. 2). The Green's function or sensitivity/model matrix (G-matrix) is derived using the meteorology of 2009 for a decay period of 59 months following 1 month of the pulsed emissions simulations. This G-matrix is repeatedly applied for the other years. Our inversion system uses quasi-interannually varying winds (IAV for simulating DACTM and cyclostationary for the basis functions) (Patra et al. 2016). The inversion has been run for 31 year (1987–2017) using 19 measurement sites (Fig. 2) at once to estimate monthly mean CH4 emission corrections to the 53 basis regions. The results are analyzed for 1988–2016 to avoid the “edge effect” (spin-up and spin-down years) in a matrix inversion system applying singular value decomposition.
2.5 Loss corrections in a posteriori emission
By ignoring the transport term in Eq. (1), which is resolved in the forward model simulation, global burden, emission, and loss budget can be written as
where
B is the global atmospheric burden,
E is total emission (sum of all
S),
L is the loss by chemical reactions, and
τ is the total lifetime. It is clear from this equation that
L is dependent on the
B (
L = B/τ) when the
DACTM is simulated. But the a priori
DACTM differs significantly from that is observed (Section 3.1) and thus, a biased value of
L, i.e., biased high when the model concentration is higher than the observed (e.g., 2005–2016) and vice versa. We have used the following equations for the loss corrections with respect to the observed atmospheric concentration ([CH
4]
obs) for the modeled concentrations.
where
-
Lpri – loss in the prior emission simulation,
-
[CH4]pri – prior model concentration (DACTM),
-
[CH4]obs – observed concentration (D).
We use the observations from the southern polar site Syowa (69°S, 39.6°E, 14 masl) because it can be considered as a background site due to the extreme remoteness from CH4 sources. By this correction, the global total a posteriori emissions were revised between −6 Tg yr−1 to 11 Tg yr−1 and 0 to 25 Tg yr−1 for the two ensemble members, for 2004–2016, following the a priori model and observation differences (Fig. 3). Such loss corrections will not be required for data assimilation systems that update the concentration fields as the model simulation progress (Bergamaschi et al. 2013; Bruhwiler et al. 2014).
2.6 Box model for δ13C-CH4
A two-box model (Turner et al. 2017) has been applied to simulate hemispheric [CH4] concentration and δ13C-CH4. The following continuity equations have solved:
The subscripts
NH and
SH denote the northern and southern hemispheres. The first term (
E) represents the emission in the respective hemisphere, the second term (
kOH [
OH][
C]) represents loss due to
OH, and the last term (
kIH ([
C]
NH − [
C]
SH)) represents the transport between both hemispheres. [
C] represents the hemispheric CH
4 concentration (
12C-CH
4) and
13C-CH
4. The
δ13C-CH
4 values are calculated using
12C-CH
4 and
13C-CH
4 simulations (
Turner et al. 2017).
The global atmospheric δ13C-CH4 is calculated from the isotopic signature of the global emission E (δ13CE) and kinetic isotope effect associated with CH4 loss processes  . The individual emission categories (Ei) from the bottom-up emissions as discussed in Table S1 have been used to calculate δ13CE of CH4.
. The individual emission categories (Ei) from the bottom-up emissions as discussed in Table S1 have been used to calculate δ13CE of CH4.
where δ13CEi and  are the isotope source signature and kinetic isotope effect for respective emission category (Ei) and loss category (Lj) as mentioned in Table S1. The CH4 chemical losses due to OH, Cl, and O(1D) were calculated from ACTM simulation using optimize emissions from 1988 to 2016.
are the isotope source signature and kinetic isotope effect for respective emission category (Ei) and loss category (Lj) as mentioned in Table S1. The CH4 chemical losses due to OH, Cl, and O(1D) were calculated from ACTM simulation using optimize emissions from 1988 to 2016.
The model was run at annual time intervals. The timescale of interhemispheric transport has been considered to be 1 year along with the global mean concentration of OH ∼ 1 × 106 molec cm−3. Table S1 provides δ13C signatures of sources and isotope fractionation factors for sink processes. Along with the prior emission, one additional case has been prepared after modifying the emissions of the industrial production (PRO; Table S1; fugitive emission) sector to obtain the best model representation for the observed changes.
2.7 Sensitivity simulations for observation network and OH variability
The control inversion uses only a limited number of sites (19) since only these stations cover the full record of atmospheric CH4 measurements for 1988–2017. We conducted another set of inversion using 16 additional observation sites, i.e., a network of 35 sites (green circles in Fig. 2), for a shorter period (1999–2017). The inversion results for this shorter period are contributed to the latest GCP-CH4 assessment (Saunois et al. 2020). The constraint on regional emission trends by inversion using 35 sites are consistent with those derived from the control inversion for the overlapping period (results are discussed in Section 3.6).
For sensitivity simulation, it is challenging to select an OH trend and variability case from the existing studies, besides the control case of an annually repeating monthly mean OH field. Figure 4 shows the assessment of OH interannual variability and trends. The magnitude OH variability in the troposphere is mostly derived from inversion of CH3CCl3 (Montzka et al. 2011; Rigby et al. 2017; Turner et al. 2017; Naus et al. 2019) or using the coupled CCMs (Dalsøren et al. 2016; He et al. 2020), and a data-driven model (Nicely et al. 2018). Most of these studies suggest an increase in OH during 1995–2005, reaching up to a value of +8 %, relative to 1988–1995 (Fig. 4). In the period after 2005, the CH3CCl3 inversion-based studies suggest a decrease in OH except for Naus et al. (2019). By contrast, CCMs suggest high levels of OH during the post-2005 period except for a dip in 2014–2015, coinciding with the most recent El Niño event. The only data-driven model suggested no significant trends for the period of our analysis, consistent with our control case for decadal mean CH4 budget (Nicely et al. 2018). As a sensitivity test (referred to as OHiav), we have chosen their “likely case” from Turner et al. (2017). Although this variability and trends in OH chosen for this sensitivity analysis could be unphysical, we only use it to illustrate the potential impact of OH on CH4 budget and concentration.

3. Results and discussion
3.1 Time evolution of global CH4 concentration and emissions
Figures 5a and 5b show the southern hemispheric (SH) mean atmospheric CH4 concentrations and growth rate (d[CH4]/dt), respectively, at annual time intervals (Section 2.3). The mean growth rate in the SH represents the global mean reasonably well (Fig. S3; Pearson's moment correlation coefficient r = 0.89). Because the northern and tropical sites exhibit a considerable site-to-site difference in CH4 growth rates and seasonality (Fig. S4), those sites are not included in Figs. 5a and 5b. The inter-site differences (ref. Fig. S4) arise from the non-linear interactions among the heterogeneity of regional emission distributions, atmospheric exposure to OH, and atmospheric transport.

The observed mean concentration and decreasing growth rate of CH4 are captured well by a priori simulations for 1988–2002 (Figs. 5a, b for SH mean and Fig. S4 for individual sites), which suggest that our knowledge of a priori CH4 emissions is consistent with the atmospheric data. This consistency in the a priori emission estimate was achieved by the use of the new anthropogenic emission inventory data from EDGARv4.3.2 (Janssens-Maenhout et al. 2019), wetland and rice emissions from VISIT land biosphere model with improved representation of the inundation area in the VISIT model (Ito et al. 2019), and a remote sensing based biomass burning emissions (GFEDv4s) (van der Werf et al. 2017).
Based on the observation and model comparison (Fig. 5b) and EDGARv4.3.2 inventory emissions (Fig. 5c, broken line), the decreasing growth rate is mostly attributed to the reduction in fugitive emissions (Fig. 5d, black line), from 95 Tg yr−1 to 92 Tg yr−1 during 1988–1991 and from 95 Tg yr−1 to 85 Tg yr−1 during 1996–1999 (Fig. 5d; see also Fig. 1), as previously inferred from atmospheric measurements (Aydin et al. 2011; Dlugokencky et al. 2003). The EDGARv4.3.2 inventory emissions also show a slight reduction (1.4 Tg yr−1) globally from enteric fermentation and manure management during 1995–1998. This finding is consistent with the magnitudes of reductions in fugitive emissions; e.g., 10–21 Tg yr−1 from 1984–2010 based on an ethane (C2H6)-CH4 joint analysis (Simpson et al. 2012) and ∼ 10 Tg yr−1 in the early 1990s after the collapse of Soviet Union (Dlugokencky et al. 2003), although the timing of the reductions varies significantly between the studies.
Superimposed on the decreasing trend during 1988–2000, the CH4 growth rate shows large interannual variability (IAV). Both the Mt. Pinatubo eruption and strong El Niño event were responsible to a great extent for the large IAV during 1991–1993, with an emission peak in 1991 likely due to the biomass burning induced by El Niño–Southern Oscillation (ENSO) (Bândâ et al. 2015; Dlugokencky et al. 1996). The a priori simulations underestimate the observed peak (∼ 14 ppb yr−1) in 1991 because of the absence of IAV in the biomass burning emissions for 1988–1996 (Fig. 5d). The inverse model suggests a ∼ 12 Tg yr−1 increase in global CH4 emissions from 1990 to 1991 (Fig. 5c). In the following years (1992–1993), a substantial decrease in the CH4 growth rate (by ∼ 12 ppb yr−1) was observed. The VISIT model suggests an 18–20 Tg reduction in global wetland CH4 emission in 1992 relative to 1988–1991 average emission (Fig. 5d). Cooler land surface temperatures, due to volcanic ash and sulfur dioxide (SO2), led to a reduction in wetland CH4 emissions from the inundated ecosystems (Ito and Inatomi 2012).
A considerable increase (∼ 8 ppb) and a decrease (∼ 10 ppb) in the CH4 growth rate is observed during the 1996–1998 El Niño and the 1998–2000 La Niña, respectively (Fig. 5b). It is also shown that the positive CH4 anomalies during positive ENSO phases are coincident with the enhanced biomass burning emissions (Simpson et al. 2006; Worden et al. 2017). For the 1997–1998 El Niño event, a fire emission anomaly of ∼ 20 Tg yr−1 is estimated by GFEDv4s inventory, whereas the VISIT wetland emissions showed an anomalous decrease in 1997, followed by increased emissions in 1998–2000 due to the La Niña conditions overall. Note also that the temperaturedependent loss of CH4 by reaction with OH in the troposphere also increases during the El Niño years (Patra et al. 2011). IAV in OH, which is not considered here, would further complicate the issue of linking the CH4 concentration anomalies with the various emission processes.
Significant discrepancies in the model–observation comparison are seen for 2002–2014 (Fig. 5a) when the MIROC4-ACTM a priori simulation overestimates the observed CH4 growth rate by ∼ 5 ppb yr−1 (Fig. 5b). This is presumably due to an overestimation of emissions from fossil fuels in EDGARv4.3.2 and its earlier releases, mainly from the coal mining (fugitive) in China (Patra et al. 2016; Peng et al. 2016; Saeki and Patra 2017; Thompson et al. 2015) (Figs. 1f, S5). The rapid increase in CH4 emissions after 2002 is mainly driven by fugitive emissions (Fig. 5d), which is debated strongly for the conterminous US (Bruhwiler et al. 2017; Sheng et al. 2018; Lan et al. 2019). The previous version of EDGAR inventory (EDGARv4.2FT) estimated a rapid increase of 27 Tg yr−1 (from 89 to 116 Tg yr−1) in global fugitive emissions during 2002–2008, whereas the net change is reduced to 19 Tg yr−1 (from 85 to 104 Tg yr−1) in the new version (EDGARv 4.3.2) by using the revised emission factors for coal mining. The issue of the emission increase rate in China will be addressed in detail later using region-specific estimates.
Table 1 shows the changes in the global burdens, the imbalance between CH4 emissions and sinks, as simulated by MIROC4-ACTM. The major sink of CH4, by reaction with OH, is determined by the changes in the CH4 burden, OH concentration, and temperature-dependent reaction rates. The control OH concentrations are taken from a three-dimensional empirical model driven by precursor observation (Spivakovsky et al. 2000). Note that no IAV in OH concentration is considered in our control simulation, and we did not address CH4 loss by reaction with atomic chlorine (Cl) in the troposphere because of the lack of sufficient information on the abundance of Cl. The chemical loss of CH4 monotonically increased from the 1990s (491 Tg yr−1 in 1990 to 508 Tg yr−1 in 1999), with relative stabilization during the 2000s (508–513 Tg yr−1) and resumed increase during 2010–2016 (516–536 Tg yr−1) with the growth rate higher than 1990s. A relatively close balance between mean CH4 emissions and sinks during 1999–2006 led to the CH4 quasi-stationary state; however, during 2007–2016, the CH4 emissions outweighed sinks and subsequently caused the renewed methane growth rate (Table S1).
3.2 Validation of global emissions using independent aircraft measurements
The a posteriori emissions are validated using an independent set of Tohoku University aircraft observations over Japan (at 0–4 km over Sendai and at 4–8 km between Sendai and Fukuoka/Sapporo) during 1988–2016 (Fig. 6). The observations were made downstream of continental East Asia and cover the whole period of our inversions (Umezawa et al. 2014). Here, we use this model–observation comparison for the overall validity of the inversion results for simulating the growth rate variability and vertical gradients. The simulated CH4 concentrations using a posteriori emission agree with the measured values mostly within ±15 ppb for individual years throughout the troposphere (Fig. 6c). The observed decline in the growth rate through 1988–1999, the quasi-stationary state in 1999–2006, and the regrowth after 2007 are reproduced well by the a posteriori simulations (Fig. 6f) at all height range (0–8 km), in contrast to the case for the simulations using a priori emissions (Fig. 6e). The correlation coefficient (r) between the observed and modeled growth rate has improved from 0.25 for a priori to 0.6 for a posteriori case. This overall agreement in CH4 for the a posteriori MIROC4-ACTM simulation with the inversion-independent observations also suggest conformity of the loss correction to the a posteriori emissions based on a priori–a posteriori loss mismatches (Section 2.5).

The source types of CH4 emissions consist of different isotopic signatures; the biogenic emissions are characterized by lower δ13C-CH4, whereas thermogenic and pyrogenic emissions are characterized by higher δ13C-CH4 relative to the values of the present-day atmosphere. We have applied a two-box model simulation of δ13C-CH4 to examine the consistency of the global emission trends based on the a priori and a posteriori emission with the observed δ13C-CH4 time series (ref. Section 2.6). The measurements of δ13CCH4 are taken from the Portland State University representative of the NH (Rice et al. 2016), 10 NOAA cooperative sites (White et al. 2019), and Ny-Ålesund by the National Institute of Polar Research (Morimoto et al. 2017; Fujita et al. 2020). The observations from different institutes are scale adjusted based on Umezawa et al. (2018) (see Fig. 7).

Using the emission sectors as per the a priori emissions, our model simulates well (within ∼ 0.05 ‰) the observed increase in δ13C-CH4 during 1988–1998 and the quasi-stationary state during 1999–2006 but failed to reproduce the fast decrease after 2008 (Fig. 7). Because of the uncertainties in representative δ13C-CH4 signatures, it is difficult to pinpoint a unique solution for a combination of emission magnitudes of various source sectors (Sherwood et al. 2017; Nisbet et al. 2019; Schwietzke et al. 2016). Instead, we adjust the global fugitive emissions trend from EDGARv4.3.2 (a priori case) by a more recent emission inventory for mainland China, which is available only during 2002–2010 (Peng et al. 2016), and further reduce the fugitive emissions gradually from 88.6 Tg yr−1 in 2011 to 83.5 Tg yr−1 in 2016 (a case similar to Schwietzke et al. (2016).
A better model–observation agreement in δ13C-CH4 decrease (Fig. 7) for the revised case suggests that the renewed CH4 growth rate is caused by an increase in fugitive emissions during 2002–2010 (by ∼ 10.9 Tg yr−1), whereas this emission increase is overestimated by the EDGARv4.3.2 inventory (at ∼ 20.8 Tg yr−1). Almost an equal amount of emission increase (∼ 10.4 Tg yr−1) is estimated from enteric fermentation by the inventories for 2002–2010. It should be mentioned here that, if an increase in OH is assumed since around 2000 as suggested by the CCMs and CH3CCl3 box models (Fig. 4), atmospheric δ13C-CH4 would show an increase after 2007, without a much greater increase in the biogenic emissions. This is not consistent with a scenario with the increasing fugitive emission compensated by enhanced OH loss, since the δ13C-CH4 observations show neither increasing nor decreasing trends for the 2000s, as recently inferred by Thompson et al. (2018).
3.4 Sensitivity to OH IAV
As no IAV in OH is considered in our control simulation, we conducted a sensitivity simulation (OHiav), where the global mean OH is varied as per Turner et al. (2017); and increased by approximately 7 % for 1988–2001 and returned close to the control value by 2007 (ref. Fig. 4). The OHiav case shows a faster decrease and increase in the CH4 growth rates than observed during 1997–2001 and 2001–2006, respectively, with an unrealistically low value of −10 ppb yr−1 in 2001 (Fig. 5b); this suggests an inconsistency of OHiav with our a priori emission cases in a 3-D chemistry–transport model framework. The a posteriori emission corresponds to the OHiav simulations (a posteriori OHiav emission) produce contrasting trends and variability in comparison with those using the control OH (Fig. 5c). The a posteriori OHiav emission shows a decreasing trend during 1998–2009, which does not reconcile well with the current knowledge of increasing emission trend from coal mining mainly in China and intensification of ruminant farming in tropical regions for the same period (Peng et al. 2016; Wolf et al. 2017). A compensation of these emission increases by a decrease in other emission sectors is possible. We further explore the spatial gradients in simulated and observed CH4 concentration in relation to the spatially explicit emission–loss budgets to extract information possible of OH-CH4-transport coupled processes. Further details of OH sensitivity on semi-hemispheric and regional emissions estimation will be addressed below.
3.5 Inter-Site Difference (ISD) in CH4: roles of emission, loss, and transport
To gain insights into the major driver for the longterm trend in the atmospheric CH4 during 1988–2016, we used the inter-site gradient (ISD) or the difference in CH4 concentration between the four target sites (GMI, SMO, KEY, and ALT) and a reference site (SYO) (ref. Fig. 2).
a. Meridional distribution of CH4 emission, loss, and burden
Figure 8 shows the target sites are located to uniquely experience the effects of emission, loss, and transport of CH4, whereas the reference site is located away from all emissions and most of the losses in either season (January or July). The tropical sites GMI and SMO are representative of the region where the dominant fraction of the OH loss occurs (approximately 60 % of global CH4 loss occurs in the region of 20°S–20°N, Figs. 8b, d). The extratropical site KEY represents the latitudinal region where emission contributions to concentration changes become dominant over those by the OH loss. The variabilities at NH high latitude site ALT keep track of the emission changes in the mid- to high latitudes, because the OH loss in the latitudes poleward of 50°N is almost negligible (Figs. 8a, c). The meridional differences of the balance between the CH4 emission and loss would appear in the observed latitudinal gradient of atmospheric CH4 from 1988 to 2016.
b. Time evolution of CH4 ISDs
The ISD between two polar sites (ALT-SYO) is observed to be greatest at 133.1 ± 5.6 ppb when averaged for 1988–2016 (the spread denotes 1-σ IAV). Mean ISD decreases sharply as the target site moved southward to the NH tropical latitudes, e.g., a value of 58.6 ± 4.5 ppb is observed for GMI-SYO (Fig. 9a). By contrast, the observed ISD of SMO-SYO is 7 ± 2 ppb; the relatively small IAV suggests that CH4 concentration at SH latitudes poleward of approximately 14°S has stayed relatively uniform in time (Fig. 9a). The a priori model simulated well the SMO-SYO CH4 ISD (4.7 ± 1.8 ppb) within their IAV, whereas greater systematic offsets are seen for the NH sites after 2005. The good model–observation agreements for SMOSYO ISDs suggest minor or no roles of CH4 emission and loss variabilities on CH4 in latitudes south of SMO, consistent with the fact that strong disparities in emission and loss rates reside mostly in the latitudes north of SMO (Figs. 8a, c, S6).

In the latitudes poleward of 30°N, CH4 emissions constitute a relatively high fraction (30 %) of the global total than that for the loss fraction, which is 10 % of the global total (Figs. 8b, d). Thus, we infer that the CH4 concentration variations at ALT are mostly contributed by the transport of emission signals from the northern mid-high latitude regions. By contrast, the fraction of loss exceeds that of the emission in the south of 15°N latitude region (Figs. 8a, c), which results in a much sharper decrease in CH4 ISD at the rate of approximately 2 ppb per latitude between KEY and SMO, compared with that between ALT and KEY (ISD decrease rate less than 1 ppb per latitude). The sharper KEY-SMO ISD is also aided by the inhibition of species mass exchange across the inter-tropical convergence zone, located around 7°N (Patra et al. 2014).
Although an average decreasing trend of the observed ALT-SYO ISD was calculated to be −0.37 ppb yr−1 for the whole period of 1988–2016, we see a substantially faster decrease during 1988–2002 (−1.51 ± 0.1 ppb yr−1) but a rapid increase during 2010–2016 (1.54 ± 0.2 ppb yr−1), following a period (2003–2009) of negligible trend (−0.06 ppb yr−1) (Fig. 9a). The decrease in the ALT-SYO CH4 from 140 to 122 ppb during 1988–2002 is coincident with the reduction in the global growth rate (Fig. S3), suggesting a common driver for both observables (Dlugokencky et al. 2003). The MIROC4-ACTM simulations using both a priori and a posteriori emission successfully simulate the ISD reduction in response to the decrease of the anthropogenic emissions in the 30–90°N latitude band during 1988–2002 (Fig. 9d).
Interestingly, the ALT-SYO CH4 ISD during 2003–2016 is lower than that during 1988–1996, despite that the global total emission is higher in the later period. According to the inversion results, this is explained by the decadal latitudinal shift in the emission agglomeration region from NH mid- and high latitudes to the tropical regions (Figs. 9d, S6d). The average ratio of a posteriori emissions at 30–60°N to those at 0–30°N was higher than 0.8 before 1996 but decreased to 0.75 during 2003–2016, whereas the emissions from the southern latitudes (10–40°S) increased monotonically during 1988–2016 (Fig. 9d). These changes could be linked to the dominated fugitive and waste emission increases in the northern extratropics, whereas the increases in ruminant farming sectors are found mainly in the tropical region (Fig. 1).
c. Evaluation of meridional (and regional) emission distributions using XCH4
To further confirm the validity of the a priori to a posteriori emission changes over the northern high latitudes, we used independent observations of total column methane (XCH4) by the GOSAT since 2009. The observations show spectacular spatial gradients in XCH4 with higher values over the intense emission regions (Figs. 10b, 1). The higher values in the tropical latitudes at a given longitude band is a result of greater tropospheric air mass (higher tropopause height), which convolved with the emission signal. Within the same latitude band, the concentration gradients are arising from orography (e.g., the contrast between northern India and Tibetan plateau) or regional emissions (e.g., eastern vs western China or conterminous US) (ref. Fig. 10b). The effect of high orography on XCH4 is identical to that of the lower tropopause height in the high latitudes.
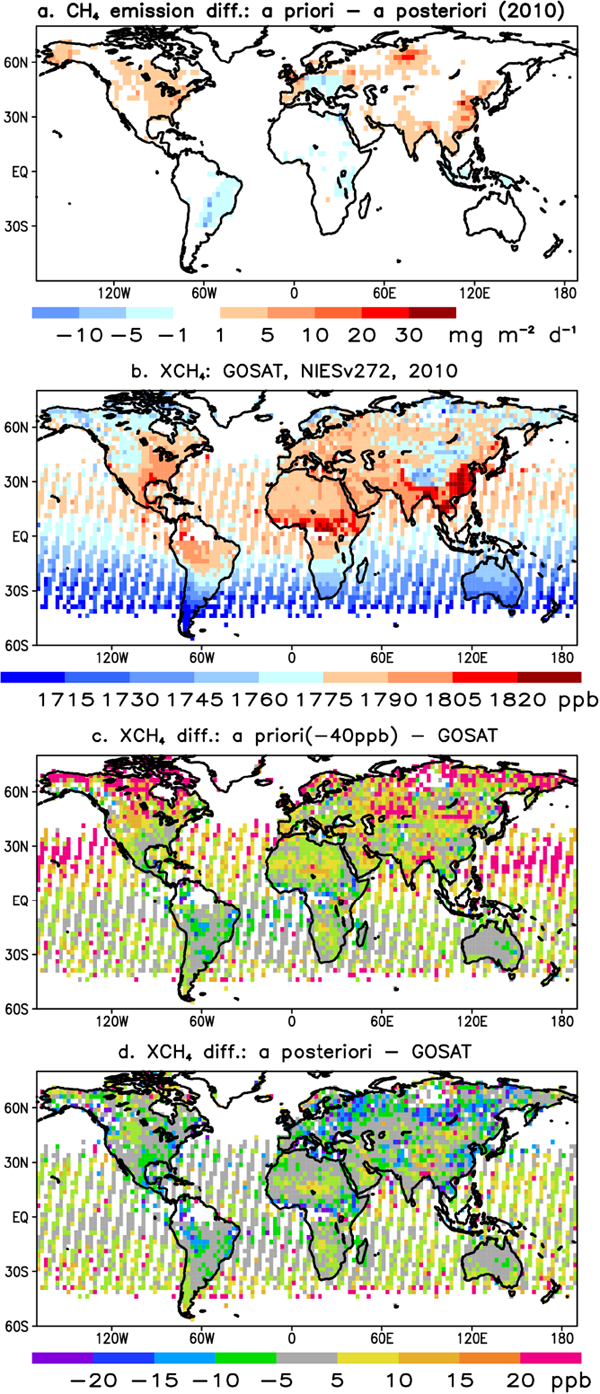
We take advantage of the global coverage to test the correctness of systematic CH4 emission decrease by the inversions in the latitude bands 30–90°N (Figs. 10a, 9d). It can also be noticed that the large (> 20 ppb) systematic XCH4 differences for the a priori model–GOSAT (Figs. 10c, d) in the high latitudes poleward of 45°N over the land, and in the north Pacific Ocean (Fig. 10c). These model–observation XCH4 differences are reduced to a range of ±10 ppb for most grids in the case of a posteriori model–GOSAT observations (Fig. 10d). The inversion using only 19 surface sites globally improves the agreement between a posteriori model simulation with independent GOSAT data. Such agreements between the observed and model XCH4 are possible with an accurate representation of the vertical distribution of CH4 between the Earth's surface and the top of the atmosphere (Houweling et al. 2017; Fujita et al. 2018).
d. Sensitivity of CH4 ISDs to loss and transport processes in the troposphere
As the loss increase is proportional to the burden, the effect of the increase of the tropical emissions on ISD would be offset by the loss in the region. The CH4 loss tendency (up to 25 ppb yr−1) is much greater than the advection tendency (±5 ppb yr−1) across 30° latitude in both hemispheres, implying more of the CH4 emitted in the tropics will be lost chemically within before they are transported to higher latitudes (Fig. 11). Thus, ISDs for all sites except ALT are relatively stable for the whole period of the analysis (1988–2016). Although the a priori model fairly simulated the observed ISDs well, it overestimates the NH ISDs for the period after 2007. These ISD increases in the a priori model after 2007 are caused by excessive emission increase in the region north of 30°N, as suggested by the a priori and a posteriori emission difference (Fig. 9d).
e. Sensitivity of CH4 ISDs to OH IAV
The simulation for the OHiav case does not show considerably different ISDs compared with those by our OH control case, particularly over the tropical sites where the loss due to OH is expected to impact CH4 concentrations the most (dotted lines in Fig. 9a). Figure 11 shows the CH4 tendencies due to the transport processes, e.g., advection, convection, and diffusion (positive and negative values show the model grids gaining or losing of CH4, respectively). The diffusion mixes CH4 emissions from the surface within the lower troposphere (mostly below 600 hPa), and then, the convection transports CH4 from the surface to the upper troposphere, whereas the advection transports CH4 meridionally at all heights, whereas the loss of CH4 by reaction with OH is occurring in the tropical latitudes mainly. The CH4 loss rate is approximately 60–80 ppb month−1 (Fig. 11a), whereas the vertical (diffusion or convection) and advection transport tendencies vary between −80 ppb month−1 and 80 ppb month−1 over the tropics (Figs. 11b–d). Therefore, any loss signal from the tropics is well mixed vertically, by which the loss signal would not be sensed strongly in the ISD at the surface sites. Despite a small difference in the ISD, the a posteriori emissions using OHiav suggest that large change (±10 Tg) can occur over 10°N–10°S compared with those over the extratropics in response to the trend in the OH concentration (dotted line in Fig. 9d).
It is noted here that this information regarding the large heterogeneity in three dimensions of the atmosphere and with time are neglected in the box modeling approaches (Naus et al. 2019), which are likely more critical for the reactive species (e.g., CH4) than the non-reactive species due to the non-linearity in loss and transport interactions. For the non-reactive species, three-dimensional features arising from their emission distribution will still be overlooked in the box modeling approaches. Our results show the interplay of dynamics and chemistry at various latitudes and heights in determining the CH4 concentration gradients.
3.6 Trends and variabilities in regional CH4 emissions
Figure 12 shows the CH4 emission anomalies in the 15 land regions for the period 1988–2016. The a posteriori emission using atmospheric data from 1) 19 sites, 2) 35 sites for a shorter period, as in the GCP-CH4 synthesis (Saunois et al. 2020), and 3) 19 sites but with the OHiav are shown for comparison. We take advantage of a joint analysis of region-specific a priori information for a few dominant sources (Fig. 1) and regional total a posteriori emission (Fig. 12) to assess the causes of the CH4 growth rates variabilities during the decrease, quasi-stationary, and regrowth phases. In general, the a posteriori emission trends agree well with a priori emission trends, except for a prominent mismatch for the East Asia region after 2002, suggesting inventory emissions are in overall agreement with the observed CH4 concentration for the control OH case. An earlier study has already shown that the inversion method successfully retrieves the regional emission anomalies from the atmospheric data even if no trends are present in the a priori emissions (Patra et al. 2016). Thus, the agreement seen here should not be interpreted as a drawback of the inversion but rather an improvement in the a priori emissions. Note also that the estimated emission anomalies using a greater number of sites (35) for a shorter duration (2000–2016) show good agreement with those using 19 sites.

Between the period 1999–2006 and 2007–2016, the a posteriori emission shows ∼ 30 Tg yr−1 increase in the global emissions (Fig. 5c, Table 1), and ∼ 24 Tg yr−1 of the global increase occurred in Asia (Fig. 12). The East Asia, West Asia, South Asia, and Southeast Asia regions accounted for 28, 19, 18, and 19 % of the global a posteriori emission increase from 1999–2006 to 2007–2016, respectively (Figs. 12f, c, h, j). The EDGARv4.3.2 inventory suggests that the coal sector over East Asia, West Asia, Southeast Asia, and ruminant farming sector over Southeast Asia drive a large part of emission increase from 1999–2006 to 2007–2016 (Figs. 5f, c, j, h). The a posteriori emission trend over East Asia shows an excursion from the continuous (extrapolated) increase of that of the a priori after 2012 (Fig. 12f). One possible explanation is that emissions from abandoned coal mines in East Asia may have ceased, in agreement with that is suggested by the δ13C-CH4 model. The most recent inventory (EDGARv5.0) emissions from coal mining indeed show a slowdown in the rate of increase, at 2.6 % yr−1 during 2012–2014 and by 3 % yr−1 during 2014–2015, relative to a rate of increase of 5.7 % yr−1 during 2003–2011 (Crippa et al. 2020).
The a posteriori emission shows 11.5 % of the global emission increase (3.2 Tg yr−1) occurred over Brazil (Fig. 12i), and 10.4 % (2.9 Tg yr−1) over Northern Africa (Fig. 12l) between 1999–2006 and 2007–2016, which is consistent with the rise in ruminant farming emission over those regions (Figs. 1i, l). The FAOSTAT statistics inferred that the meat and other cattle population over Latin America and the swine population in South and Southeast Asia increased during the 2000s (FAOSTAT 2018). Using the updated emission factors, Wolf et al. (2017) also suggested that the ruminant farming sector largely contributes to the annual CH4 emissions increase since 2007.
More recently, a posteriori emission over Temperate North America (Fig. 12e) shows a 9.8 Tg yr−1 increase (from 30 Tg yr−1 in 2010 to 39.8 Tg yr−1 in 2016) between 2010 and 2016, which is consistent with the increase in wetland emission by VISIT model and fugitive emissions in EDGARv4.3.2 inventory during the same period (Fig. 1e). This emission increase outside fast-growing economies may have helped to sustain the regrowth in the 2010s. However, the increase in emissions from fugitive sources from the United States is uncertain (Sheng et al. 2018; Lan et al. 2019), and a broader assessment is needed for the wetland emission increase.
The higher northern regions show a systematic decrease in a posteriori emission. The European emissions gradually decreased between 1988 and 2010 (Fig. 12b) due to the decline in ruminant farming emissions (Fig. 1b), and Russian emissions decreased between 1988 and 2000 (Fig. 12d) due to the decrease in fugitive emissions (Fig. 1d). The FAOSTAT statistics suggest that the cow and cattle population decreased largely over western Europe (FAOSTAT 2018). As reported by some previous studies (Sweeney et al. 2016; Thompson et al. 2018), we did not find any detectable increase in natural CH4 emissions from the wetlands and other ecosystems in the northern high latitudes, e.g., the Boreal North America and Russia, despite the increase in annual-mean temperature by ∼ 1.2°C decade−1 from 1985 to 2015 over the Arctic region (Sweeney et al. 2016). These regions are expected to release CH4 in response to future global warming in the boreal zones (Anthony et al. 2018).
A comparison between the control OH (orange line) and OHiav (purple line) cases suggest that change in OH can affect the estimation of a posteriori emission anomalies in the tropical latitudes i.e. Brazil, Central Africa, South Asia, and Southeast Asia regions (Figs. 12h–j, m). Given the a priori knowledge of human activities (e.g., ruminant farming) and no reports about significant changes in the wetland area or climate variability, it is difficult to explain the emission anomalies for the OHiav case, such as a decrease in emissions for Brazil and Central Asia (2000–2016), fast increase during 1991–1999 and stable afterward for South Asia, and a decrease for Southeast Asia (1999–2009). Other regions with high CH4 emissions and the tropical proximity region show some differences between a posteriori emission using the two OH cases. However, our conclusions regarding the timing and magnitude of emission change are relatively unaffected, e.g., the cases of East Asia, Tropical America, Temperate South America, and Northern Africa (Figs. 12f, g, k, l). Further, the higher latitude regions, where emissions are mostly responsible for the CH4 ISD changes, do not see any difference in trend due to the OH variability case examined here. We, therefore, conclude that the predominance of CH4 emissions in controlling the CH4 growth rates for the past three decades is very likely.
3.7 Comparisons of China and India CH4 emissions
China in East Asia and India in South Asia are two of the fastest-growing economies in the world and their CH4 emission trends are assessed in recent years (Bergamaschi et al. 2013; Thompson et al. 2015; Miller et al. 2019; Ganesan et al. 2017). A comparison of four different inversion results for China shows excellent agreement for the CH4 emission anomaly for 2000–2016 (Fig. 13a), which is expected because we find that the variability in OH, if any, does not affect East Asian emission inversion. Moreover, the XCH4 observation by GOSAT (Yoshida et al. 2013; Kuze et al. 2016) shows reasonable coverage in all seasons over China (Fig. 10). Notably, our results show fine agreement with the estimated country-totals and IAV in CH4 emissions using GOSAT data (Miller et al. 2019) from 2010 to 2015, except for 2011.

However, the comparison of CH4 emissions over India shows large discrepancies between our results and previous studies. The estimated emissions by Ganesan et al. (2017) and Miller et al. (2019) are 58 % (22 Tg yr−1) and 28 % (35 Tg yr−1) respectively lower than that in this study (49 Tg yr−1). We suspect that the differences arise from the modeling tools used (regional vs global models), and more importantly, the treatments of data gaps and data screening for inversion. The GOSAT has only very few retrievals during the summer/monsoon seasons because of cloud cover (Chandra et al. 2017). The quality may also be in question because the data from the summer season under the high humidity conditions show much lower values compared with our model results in all years (Fig. S7b). We are currently unable to reason the disparity in our results with Ganesan et al. (2017)—a possible explanation is that their study does not include all emission sectors as we do. This suspect stems from the good comparisons obtained between the MIROC4-ACTM simulations and GOSAT XCH4 for the zonal cross-sections over the India (also China) regions as depicted in Figs. S7c–f.
The model–model differences cannot be reconciled without further analysis involving models with common input parameters such as emissions and chemical loss. A coordinated model intercomparison is needed for better diagnostics of these model–model differences.
3.8 Separation of major controlling factors for CH4 growth phases
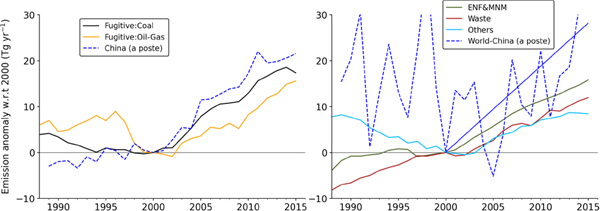
Our inverse modeling was setup using EDGARv 4.3.2 anthropogenic emission inventory. Since that was completed, a new version EDGARv5.0 has been released (Crippa et al. 2020). We compare our inversion emission anomalies for China with the fugitive emission sector of EDGARv5.0 (Fig. 14a). Approximately 80 % of the emission increase estimated for China (21.5 Tg yr−1) during 2000–2015 can be explained by the emission increase due to global fugitive coal emissions. As discussed earlier, the emission decrease from fugitive emissions from the oil and gas industry helped to stabilize CH4 concentration in the 1990s and then contributed to the renewed CH4 growth since the late 2000s. The other major sector source that drove changes in the CH4 growth rate arises from an increase in emissions from enteric fermentation and manure management, 15 Tg yr−1 during 2000–2015. This increase contributed 56 % to the emission increase from the rest of the world at 28 Tg yr−1 (Fig. 14b). Note that the increase in emissions from enteric fermentation and manure management is caused by the greater intensity of ruminant farming as estimated by the FAO and the emission inventories (Crippa et al. 2020; Wolf et al. 2017; FAOSTAT 2018). The inventory emissions from waste treatments can account for up to 43 % of the linear increase in emissions for the rest of the world. The trends in other anthropogenic emissions, as in EDGARv5.0, are balanced by the natural emission changes globally or by the changes in CH4 loss due to the reaction with OH in the troposphere. In summary, the changes in anthropogenic emissions have dominated the contributions to the three different CH4 growth rate phases from 1988 to 2016, whereas natural emissions likely dominated the IAV in CH4 emissions (ref. broken blue line in Fig. 14b).