DESCRIPTION
Decitabine
Decitabine is a nucleoside metabolic inhibitor. Decitabine is a white to off-white solid with the molecular formula of C8H12N4O4 and a molecular weight of 228.21 daltons. Its international union of pure and applied chemistry (IUPAC) chemical name is 4-amino-1-[(2R,4S,5R)-4-hydroxy-5(hydroxymethyl)oxolan-2-yl]-1,3,5-triazin-2(1H)-one and it has the following structural formula:
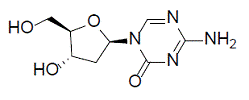 |
Cedazuridine
Cedazuridine is a cytidine deaminase inhibitor. Cedazuridine is a white to off-white solid with the molecular formula of C9H14F2N2O5 and a molecular weight of 268.21 daltons. Its IUPAC chemical name is (4R)-1-[(2R,4R,5R)-3,3-difluoro-4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-4-hydroxy-1,3-diazinan-2one and it has the following structural formula:
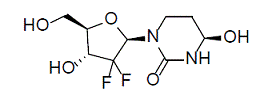 |
Inqovi
Inqovi (decitabine and cedazuridine) tablets, for oral use contain 35 mg decitabine and 100 mg cedazuridine. The tablets are biconvex, oval-shaped, film-coated, red and debossed with “H35” on one side. Each film-coated tablet contains the following inactive ingredients: lactose monohydrate, hypromellose, croscarmellose sodium, colloidal silicon dioxide, and magnesium stearate. The film coating material contains polyvinyl alcohol, titanium dioxide, polyethylene glycol, talc, and iron oxide red.
Uses for Inqovi
INQOVI is indicated for treatment of adult patients with myelodysplastic syndromes (MDS), including previously treated and untreated, de novo and secondary MDS with the following French-American- British subtypes (refractory anemia, refractory anemia with ringed sideroblasts, refractory anemia with excess blasts, and chronic myelomonocytic leukemia [CMML]) and intermediate-1, intermediate-2, and high-risk International Prognostic Scoring System groups.
Dosage for Inqovi
Important Administration Information
Do NOT substitute INQOVI for an intravenous decitabine product within a cycle.
Consider administering antiemetics prior to each dose to minimize nausea and vomiting [see ADVERSE REACTIONS].
Recommended Dosage
The recommended dosage of INQOVI is 1 tablet (containing 35 mg decitabine and 100 mg cedazuridine) orally once daily on Days 1 through 5 of each 28-day cycle for a minimum of 4 cycles until disease progression or unacceptable toxicity. A complete or partial response may take longer than 4 cycles.
Instruct patients of the following:
- Take INQOVI at the same time each day.
- Swallow tablets whole. Do not cut, crush, or chew tablets.
- Do not consume food 2 hours before and 2 hours after each dose.
- Take one tablet a day for 5 days in each cycle. If the patient misses a dose within 12 hours of the time it is usually taken, instruct patients to take the missed dose as soon as possible and then to resume the normal daily dosing schedule. Extend the dosing period by one day for every missed dose to complete 5 daily doses for each cycle.
- Do not take an additional dose if vomiting occurs after INQOVI administration but continue with the next schedule dose.
INQOVI is a hazardous drug. Follow applicable special handling and disposal procedures.1
Monitoring And Dosage Modifications For Adverse Reactions
Hematologic Adverse Reactions
Obtain complete blood cell counts prior to initiating INQOVI and before each cycle. Delay the next cycle if absolute neutrophil count (ANC) is less than 1,000/μL and platelets are less than 50,000/μL in the absence of active disease. Monitor complete blood cell counts until ANC is 1,000/μL or greater and platelets are 50,000/μL or greater [see WARNINGS AND PRECAUTIONS].
- If hematologic recovery occurs (ANC at least 1,000/μL and platelets at least 50,000/μL) within 2 weeks of achieving remission, continue INQOVI at the same dose.
- If hematologic recovery does not occur (ANC at least 1,000/μL and platelets at least 50,000/μL) within 2 weeks of achieving remission,
- Delay INQOVI for up to 2 additional weeks AND
- Resume at a reduced dose by administering INQOVI on Days 1 through 4. Consider further dose reductions in the order listed in Table 1 if myelosuppression persists after a dose reduction. Maintain or increase dose in subsequent cycles as clinically indicated.
Table 1: Recommended INQOVI Dose Reductions for Myelosuppression
| Dose Reduction | Dosage |
| First | 1 tablet orally once daily on Days 1 through 4 |
| Second | 1 tablet orally once daily on Days 1 through 3 |
| Third | 1 tablet orally once daily on Days 1, 3 and 5 |
Manage persistent severe neutropenia and febrile neutropenia with supportive treatment [see WARNINGS AND PRECAUTIONS].
Non-Hematologic Adverse Reactions
Delay the next cycle for the following non-hematologic adverse reactions and resume at the same or reduced dose upon resolution:
- Serum creatinine 2 mg/dL or greater
- Serum bilirubin 2 times upper limit of normal (ULN) or greater
- Aspartate aminotransferase (AST) or alanine aminotransferase (ALT) 2 times ULN or greater
- Active or uncontrolled infection
HOW SUPPLIED
Dosage Forms And Strengths
INQOVI tablets contain 35 mg decitabine and 100 mg cedazuridine. The tablets are biconvex, oval-shaped, film-coated, red and debossed with “H35” on one side.
INQOVI tablets are biconvex, oval-shaped, film-coated, red, and debossed with “H35” on one side.
The tablets are packaged in blisters and supplied as follows:
NDC: 64842-0727-9; 5 tablets in one blister card in a child-resistant carton
Storage And Handling
Store at 20°C to 25°C (68°F to 77°F); excursions permitted from 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature]. Dispense medication in the original packaging.
INQOVI is a hazardous drug. Follow applicable special handling and disposal procedures.1
REFERENCES
1. OSHA Hazardous Drugs. OSHA. http://www.osha.gov/SLTC/hazardousdrugs/index.html
Manufactured for: Otsuka Pharmaceutical Co., Ltd. Japan. Distributed by: Taiho Oncology, Inc., Princeton, NJ 08540 USA. Revised: Mar 2022
Side Effects for Inqovi
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Myelosuppression [see WARNINGS AND PRECAUTIONS]
Clinical Trials Experience
Because clinical trials are conducted under widely variable conditions, adverse event rates observed in clinical trials of a drug cannot be directly compared with rates of clinical trials of another drug and may not reflect the rates observed in practice.
Myelodysplastic Syndrome And Chronic Myelomonocytic Leukemia
The safety of INQOVI was evaluated in a pooled safety population that includes patients enrolled in Study ASTX727-01-B and Study ASTX727-02 [see Clinical Studies].
Patients were randomized to receive INQOVI (35 mg decitabine and 100 mg cedazuridine) orally once daily on Days 1 through 5 in Cycle 1 and decitabine 20 mg/m² intravenously on Days 1 through 5 in Cycle 2, or the reverse sequence, and then INQOVI (35 mg decitabine and 100 mg cedazuridine) orally once daily on Days 1 through 5 of each 28-day cycle in Cycles 3 and beyond. Patients were allowed to have one prior cycle of decitabine or azacitidine and there was no limit for body weight or surface area. Among the patients who received INQOVI, 61% of patients were exposed for 6 months or longer and 24% were exposed to INQOVI for greater than 1 year.
Serious adverse reactions occurred in 68% of patients who received INQOVI. Serious adverse reactions in > 5% of patients included febrile neutropenia (30%), pneumonia (14%), and sepsis (13%). Fatal adverse reactions occurred in 6% of patients. These included sepsis (1%), septic shock (1%), pneumonia (1%), respiratory failure (1%), and one case each of cerebral hemorrhage and sudden death.
Permanent discontinuation due to an adverse reaction occurred in 5% of patients who received INQOVI. The most frequent adverse reactions resulting in permanent discontinuation were febrile neutropenia (1%) and pneumonia (1%).
Dose interruptions due to an adverse reaction occurred in 41% of patients who received INQOVI. Adverse reactions requiring dosage interruptions in > 5% of patients who received INQOVI included neutropenia (18%), febrile neutropenia (8%), thrombocytopenia (6%), and anemia (5%).
Dose reductions due to an adverse reaction occurred in 19% of patients who received INQOVI. Adverse reactions requiring dosage reductions in > 2% of patients who received INQOVI included neutropenia (12%), anemia (3%), and thrombocytopenia (3%).
The most common adverse reactions (≥ 20%) were fatigue, constipation, hemorrhage, myalgia, mucositis, arthralgia, nausea, dyspnea, diarrhea, rash, dizziness, febrile neutropenia, edema, headache, cough, decreased appetite, upper respiratory tract infection, pneumonia, and transaminase increased. The most common Grade 3 or 4 laboratory abnormalities (≥ 50%) were leukocytes decreased, platelet count decreased, neutrophil count decreased, and hemoglobin decreased.
Table 2 summarizes the adverse reactions in the pooled safety population.
Table 2: Adverse Reactions (≥ 10%) in Patients Who Received INQOVI in Pooled Safety Population
| Adverse Reactions | INQOVI Cycle 1 N=107 |
Intravenous Decitabine Cycle 1 N=106 |
INQOVIf All Cycles N=208 |
|||
| All Grades (%) | Grades 3-4 (%) | All Grades (%) | Grades 3-4 (%) | All Grades (%) | Grades 3-4 (%) | |
| General disorders and administration site conditions | ||||||
| Fatigue1 | 29 | 2 | 25 | 0 | 55 | 5 |
| Hemorrhage2 | 24 | 2 | 17 | 0 | 43 | 3 |
| Edema3 | 10 | 0 | 11 | 0 | 30 | 0.5 |
| Pyrexia | 7 | 0 | 7 | 0 | 19 | 1 |
| Gastrointestinal disorders | ||||||
| Constipation4 | 20 | 0 | 23 | 0 | 44 | 0 |
| Mucositis5 | 18 | 1 | 24 | 2 | 41 | 4 |
| Nausea | 25 | 0 | 16 | 0 | 40 | 0.5 |
| Diarrhea6 | 16 | 0 | 11 | 0 | 37 | 1 |
| Transaminase increased7 | 12 | 1 | 3 | 0 | 21 | 3 |
| Abdominal pain8 | 9 | 0 | 7 | 0 | 19 | 1 |
| Vomiting | 5 | 0 | 5 | 0 | 15 | 0 |
| Musculoskeletal and connective tissue disorders | ||||||
| Myalgia9 | 9 | 2 | 16 | 1 | 42 | 3 |
| Arthralgia10 | 9 | 1 | 13 | 1 | 40 | 3 |
| Respiratory, thoracic, and mediastinal disorders | ||||||
| Dyspnea11 | 17 | 3 | 9 | 3 | 38 | 6 |
| Cough12 | 7 | 0 | 8 | 0 | 28 | 0 |
| Blood & lymphatic system disorders | ||||||
| Febrile neutropenia | 10 | 10 | 13 | 13 | 33 | 32 |
| Skin and subcutaneous tissue disorders | ||||||
| Rash13 | 12 | 1 | 11 | 1 | 33 | 0.5 |
| Nervous system disorders | ||||||
| Dizziness14 | 16 | 1 | 11 | 0 | 33 | 2 |
| Headache15 | 22 | 0 | 13 | 0 | 30 | 0 |
| Neuropathy16 | 4 | 0 | 8 | 0 | 13 | 0 |
| Metabolism and nutritional disorders | ||||||
| Decreased appetite | 10 | 1 | 6 | 0 | 24 | 2 |
| Infections and infestations | ||||||
| Upper respiratory tract infection17 | 6 | 0 | 3 | 0 | 23 | 1 |
| Pneumonia18 | 7 | 7 | 7 | 5 | 21 | 15 |
| Sepsis19 | 6 | 6 | 2 | 1 | 14 | 11 |
| Cellulitis20 | 4 | 1 | 3 | 2 | 12 | 5 |
| Investigations | ||||||
| Renal impairment21 | 9 | 0 | 8 | 1 | 18 | 0 |
| Weight decreased | 5 | 0 | 3 | 0 | 10 | 1 |
| Injury, poisoning, and procedural complications | ||||||
| Fall | 4 | 0 | 1 | 0 | 12 | 1 |
| Psychiatric disorders | ||||||
| Insomnia | 6 | 0 | 2 | 0 | 12 | 0.5 |
| Vascular disorders | ||||||
| Hypotension22 | 4 | 0 | 6 | 1 | 11 | 2 |
| Cardiac Disorders | ||||||
| Arrhythmia23 | 3 | 0 | 2 | 0 | 11 | 1 |
| †Includes adverse reactions that occurred during all cycles, including during treatment with 1 cycle of intravenous decitabine. 1 Includes fatigue, asthenia, and lethargy 2 Includes contusion, epistaxis, petechiae, hematuria, conjunctival hemorrhage, mouth hemorrhage, purpura, angina bullosa hemorrhagica, gingival bleeding, hematoma, hemoptysis, eye contusion, hemorrhagic diathesis, increased tendency to bruise, vaginal hemorrhage, abdominal wall hematoma, blood blister, bone contusion, catheter site bruise, ecchymosis, genital hemorrhage, intra- abdominal hematoma, oral mucosa hematoma, periorbital hemorrhage, procedural hemorrhage, pulmonary alveolar hemorrhage, retinal hemorrhage, scleral hemorrhage, thrombotic thrombocytopenic purpura, tongue hemorrhage, and vessel puncture site hemorrhage 3 Includes edema peripheral, peripheral swelling, swelling face, fluid overload, localized edema, face edema, edema, eye swelling, eyelid edema, fluid retention, periorbital swelling, scrotal edema, scrotal swelling, and swelling 4 Includes constipation and feces hard 5 Includes oropharyngeal pain, stomatitis, mouth ulceration, proctalgia, oral pain, gingivitis, oral disorder, gingival pain, colitis, glossodynia, mouth swelling, pharyngitis, proctitis, duodenitis, enteritis, gingival discomfort, gingival swelling, lip disorder, lip ulceration, mucosal ulceration, nasal ulcer, noninfective gingivitis, oral mucosal blistering, oral mucosal erythema, pharyngeal erythema, pharyngeal ulceration, tongue ulceration, and vulvitis 6 Includes diarrhea and feces soft 7 Includes alanine aminotransferase increased, aspartate aminotransferase increased, blood alkaline phosphatase increased, gammaglutamyltransferase increased, liver function test increased, and transaminases increased 8 Includes abdominal pain, abdominal pain upper, abdominal pain lower, epigastric discomfort, and abdominal discomfort 9 Includes myalgia, pain in extremity, muscle spasms, pain, musculoskeletal pain, non-cardiac chest pain, muscular weakness, musculoskeletal chest pain, flank pain, musculoskeletal stiffness, muscle strain, and musculoskeletal discomfort 10 Includes arthralgia, back pain, neck pain, joint stiffness, pain in jaw, joint swelling, bursitis, joint range of otion decreased, and joint injury 11 Includes dyspnea, dyspnea exertional, hypoxia, wheezing, chronic obstructive pulmonary disease, and tachypnoea 12 Includes cough and productive cough 13 Includes maculo-papular rash, rash, erythema, skin lesion, folliculitis, dermatitis, dermatitis acneiform, eczema, erythema multiforme, rash erythematous, seborrheic keratosis, skin ulcer, dermatitis allergic, dermatitis contact, eczema nummular, genital erythema, rash papular, rash pruritic, rash pustular, seborrheic dermatitis, skin exfoliation, skin irritation, stasis dermatitis, and ulcerative keratitis 14 Includes dizziness, vertigo, postural dizziness, and positional vertigo 15 Includes headache, sinus pain, and sinus headache 16 Includes hypoesthesia, paresthesia, neuropathy peripheral, gait disturbance, peripheral sensory neuropathy, ataxia, balance disorder, brachial plexopathy, carpal tunnel syndrome, and radicular pain 17 Includes upper respiratory tract infection, nasopharyngitis, sinusitis, and viral upper respiratory tract infection 18 Includes pneumonia, pneumonitis, atypical pneumonia, and lung infection 19 Includes sepsis, bacteremia, septic shock, endocarditis, pseudomonal bacteremia, and staphylococcal bacteremia 20 Includes cellulitis, catheter site cellulitis, and infected bite 21 Includes blood creatinine increased, acute kidney injury, blood urea increased, blood creatine increased, and renal failure 22 Includes hypotension, blood pressure decreased, and cardiogenic shock 23 Includes sinus tachycardia, atrial fibrillation, bradycardia, tachycardia, atrial flutter, sinus bradycardia, and conduction disorder |
||||||
Clinically relevant adverse reactions in < 10% of patients who received INQOVI included:
- Acute febrile neutrophilic dermatosis (Sweet's syndrome) (1%)
- Tumor lysis syndrome (0.5%)
Table 3: Select Laboratory Abnormalities (> 20%) Worsening from Baseline in Patients Who Received INQOVI in Pooled Safety Population
| Lab Abnormality* | INQOVI Cycle 1† | Intravenous Decitabine Cycle 1† | INQOVI All Cycles† | |||
| All Grades (%) | Grades 3-4 (%) | All Grades (%) | Grades 3-4 (%) | All Grades (%) | Grades 3-4 (%) | |
| Hematology | ||||||
| Leukocytes decreased | 79 | 65 | 77 | 59 | 87 | 81 |
| Platelet count decreased | 79 | 65 | 77 | 67 | 82 | 76 |
| Neutrophil count decreased | 70 | 65 | 62 | 59 | 73 | 71 |
| Hemoglobin decreased | 58 | 41 | 59 | 36 | 71 | 55 |
| Chemistry | ||||||
| Glucose increased | 19 | 0 | 11 | 0 | 54 | 7 |
| Albumin decreased | 22 | 1 | 20 | 0 | 45 | 2 |
| Alkaline phosphatase increased | 22 | 1 | 12 | 0 | 42 | 0.5 |
| Glucose decreased | 14 | 0 | 17 | 0 | 40 | 1 |
| Alanine aminotransferase increased | 13 | 1 | 7 | 0 | 37 | 2 |
| Sodium decreased | 9 | 2 | 8 | 0 | 30 | 4 |
| Calcium decreased | 16 | 0 | 12 | 0 | 30 | 2 |
| Aspartate aminotransferase increased | 6 | 1 | 2 | 0 | 30 | 2 |
| Creatinine increased | 7 | 0 | 8 | 0 | 29 | 0.5 |
| * Includes any lab abnormalities that worsened by one or more grades. Grade 3-4 includes any lab abnormalities that worsened to Grade 3 or Grade 4. † The denominator used to calculate the rate varied from 103 to 107 for INQOVI Cycle 1, from 102 to 106 for Intravenous Decitabine Cycle and from 203 to 208 for INQOVI All Cycles based on the number of patients with a baseline value and at least one post-treatment value. |
||||||
Postmarketing Experience
The following adverse reactions have been identified during postapproval use of intravenous decitabine. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Blood and Lymphatic System Disorders: Differentiation syndrome
Respiratory, Thoracic and Mediastinal Disorders: Interstitial lung disease
Cardiac Disorders: Cardiomyopathy
Drug Interactions for Inqovi
Effects Of INQOVI On Other Drugs
Drugs Metabolized By Cytidine Deaminase
Cedazuridine is an inhibitor of the cytidine deaminase (CDA) enzyme. Coadministration of INQOVI with drugs that are metabolized by CDA may result in increased systemic exposure with potential for increased toxicity of these drugs [see CLINICAL PHARMACOLOGY]. Avoid coadministration of INQOVI with drugs that are metabolized by CDA.
Warnings for Inqovi
Included as part of the PRECAUTIONS section.
Precautions for Inqovi
Myelosuppression
Fatal and serious myelosuppression can occur with INQOVI. Based on laboratory values, new or worsening thrombocytopenia occurred in 82% of patients, with Grade 3 or 4 occurring in 76%. Neutropenia occurred in 73% of patients, with Grade 3 or 4 occurring in 71%. Anemia occurred in 71% of patients, with Grade 3 or 4 occurring in 55%. Febrile neutropenia occurred in 33% of patients, with Grade 3 or 4 occurring in 32%. Myelosuppression (thrombocytopenia, neutropenia, anemia, and febrile neutropenia) is the most frequent cause of INQOVI dose reduction or interruption, occurring in 36% of patients. Permanent discontinuation due to myelosuppression (febrile neutropenia) occurred in 1% of patients. Myelosuppression and worsening neutropenia may occur more frequently in the first or second treatment cycles and may not necessarily indicate progression of underlying MDS.
Fatal and serious infectious complications can occur with INQOVI. Pneumonia occurred in 21% of patients, with Grade 3 or 4 occurring in 15%. Sepsis occurred in 14% of patients, with Grade 3 or 4 occurring in 11%. Fatal pneumonia occurred in 1% of patients, fatal sepsis in 1%, and fatal septic shock in 1% [see ADVERSE REACTIONS].
Obtain complete blood cell counts prior to initiation of INQOVI, prior to each cycle, and as clinically indicated to monitor response and toxicity. Administer growth factors and anti-infective therapies for treatment or prophylaxis as appropriate. Delay the next cycle and resume at the same or reduced dose as recommended [see DOSAGE AND ADMINISTRATION].
Embryo-Fetal Toxicity
Based on findings from human data, animal studies, and its mechanism of action, INQOVI can cause fetal harm when administered to a pregnant woman. In nonclinical studies with decitabine in mice and rats, decitabine was teratogenic, fetotoxic, and embryotoxic at doses less than the recommended human dose.
Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with INQOVI and for 6 months after the last dose. Advise males with female partners of reproductive potential to use effective contraception during treatment with INQOVI and for 3 months after the last dose [see Use In Specific Populations].
Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (PATIENT INFORMATION).
Myelosuppression
Advise patients of the risk of myelosuppression and to report any symptoms of fever, infection, anemia, or bleeding to their healthcare provider as soon as possible. Advise patients for the need for laboratory monitoring [see WARNINGS AND PRECAUTIONS].
Embryo-Fetal Toxicity
Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to inform their healthcare provider of a known or suspected pregnancy [see WARNINGS AND PRECAUTIONS, Use In Specific Populations].
Advise females of reproductive potential to use effective contraception during treatment with INQOVI and for 6 months after the last dose [see Use In Specific Populations].
Advise males with female partners of reproductive potential to use effective contraception during treatment with INQOVI and for 3 months after the last dose [see Use In Specific Populations, Nonclinical Toxicology].
Lactation
Advise women not to breastfeed during treatment with INQOVI and for 2 weeks after the last dose [see Use In Specific Populations].
Administration
Advise patients to take INQOVI at approximately the same time each day on an empty stomach. Instruct patients to avoid eating for at least 2 hours before and 2 hours after taking INQOVI. Advise patients on what to do when a dose is missed or vomited [see DOSAGE AND ADMINISTRATION].
Nonclinical Toxicology
Carcinogenesis, Mutagenesis, Impairment Of Fertility
Carcinogenicity studies with decitabine, cedazuridine, or their combination have not been conducted.
INQOVI is genotoxic. Decitabine increased mutation frequency in L5178Y mouse lymphoma cells, and mutations were produced in an E. coli lac-I transgene in colonic DNA of decitabine-treated mice. Decitabine also caused chromosomal rearrangements in larvae of fruit flies. Cedazuridine was genotoxic in a reverse bacterial mutation assay (Ames assay) and in an in vitro chromosomal aberration study using human lymphocytes.
Fertility and repeat-dose toxicity studies in animals showed adverse outcomes on reproductive function and fertility. In male mice given intraperitoneal injections of 0.15, 0.3, or 0.45 mg/m² decitabine (approximately 0.3% to 1% the recommended clinical dose) 3 times a week for 7 weeks, testes weights were reduced, abnormal histology was observed, and significant decreases in sperm number were found at doses ≥ 0.3 mg/m². In females mated to males dosed with ≥ 0.3 mg/m² decitabine, pregnancy rate was reduced, and preimplantation loss was significantly increased.
Decitabine was administered orally to rats at 0.75, 2.5, or 7.5 mg/kg/day in cycles of 5-days-on/23-daysoff for a total of 90 days. Low testes and epididymis weights, abnormal histology, and reduced sperm number were observed at doses ≥ 0.75 mg/kg. The dose of 0.75 mg/kg resulted in exposures in animals that were approximately 3 times the exposure in patients at the recommended clinical dose based on AUC.
Cedazuridine was administered orally to mice at 100, 300, or 1,000 mg/kg/day in cycles of 7-days-on/21- days-off for a total of 91 days. Adverse findings in male and female reproductive organs were observed at the 1,000 mg/kg dose and included abnormal histology in the testes and epididymis, reduced sperm number, and abnormal histology in the ovary. The dose of 1,000 mg/kg/day resulted in exposures in animals that were approximately 108 times the exposure in patients at the recommended clinical dose. Adverse effects in male and female reproductive organs were reversible following a recovery period.
Use In Specific Populations
Pregnancy
Risk Summary
Based on findings from human data, animal studies, and its mechanism of action [see CLINICAL PHARMACOLOGY], INQOVI can cause fetal harm when administered to a pregnant woman. A single published case report of intravenous decitabine use throughout the first trimester during pregnancy describes adverse developmental outcomes, including major birth defects (structural abnormalities). In animal reproduction studies, intravenous administration of decitabine to pregnant mice and rats during organogenesis at doses approximately 7% of the recommended human dose on a body surface area (mg/m²) basis caused adverse developmental outcomes, including increased embryo-fetal mortality, alterations to growth, and structural abnormalities (see Data). Advise pregnant women of the potential risk to a fetus.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Human Data
There are no available data on INQOVI use in pregnant women.
A single published case report of intravenous decitabine pregnancy exposure in a 39-year-old woman with a hematologic malignancy described multiple structural abnormalities after 6 cycles of therapy in the 18th week of gestation. These abnormalities included holoprosencephaly, absence of nasal bone, midfacial deformity, cleft lip and palate, polydactyly, and rocker-bottom feet. The pregnancy was terminated.
Animal Data
No reproductive or developmental toxicity studies have been conducted with INQOVI or cedazuridine.
In utero exposure to decitabine causes temporal-related defects in the rat and/or mouse, which include growth suppression, exencephaly, defective skull bones, rib/sternabrae defects, phocomelia, digit defects, micrognathia, gastroschisis, and micromelia. Decitabine inhibits proliferation and increases apoptosis of neural progenitor cells of the fetal central nervous system (CNS) and induces palatal clefting in the developing murine fetus. Studies in mice have also shown that decitabine administration during osteoblastogenesis (Day 10 of gestation) induces bone loss in offspring.
In mice exposed to single intraperitoneal decitabine injections (0, 0.9 and 3.0 mg/m², approximately 2% and 7% of the recommended daily clinical dose, respectively) over gestation Days 8, 9, 10 or 11, no maternal toxicity was observed, but reduced fetal survival was observed after treatment at 3 mg/m² and decreased fetal weight was observed at both dose levels. The 3 mg/m² dose elicited characteristic fetal defects for each treatment day, including supernumerary ribs (both dose levels), fused vertebrae and ribs, cleft palate, vertebral defects, hind-limb defects, and digital defects of fore- and hind-limbs.
In rats given a single intraperitoneal injection of 2.4, 3.6 or 6 mg/m² decitabine (approximately 5, 8, or 13% the daily recommended clinical dose, respectively) on gestation Days 9-12, no maternal toxicity was observed. No live fetuses were seen at any dose when decitabine was injected on gestation Day 9. A significant decrease in fetal survival and reduced fetal weight at doses greater than 3.6 mg/m² was seen when decitabine was given on gestation Day 10. Increased incidences of vertebral and rib anomalies were seen at all dose levels, and induction of exophthalmia, exencephaly, and cleft palate were observed at 6.0 mg/m². Increased incidence of foredigit defects was seen in fetuses at doses greater than 3.6 mg/m². Reduced size and ossification of long bones of the fore-limb and hind-limb were noted at 6 mg/m².
The effect of decitabine on postnatal development and reproductive capacity was evaluated in mice administered a single 3 mg/m² intraperitoneal injection (approximately 7% the recommended daily clinical dose) on Day 10 of gestation. Body weights of males and females exposed in utero to decitabine were significantly reduced relative to controls at all postnatal time points. No consistent effect on fertility was seen when female mice exposed in utero were mated to untreated males. Untreated females mated to males exposed in utero showed decreased fertility at 3 and 5 months of age (36% and 0% pregnancy rate, respectively). Follow up studies indicated that treatment of pregnant mice with decitabine on gestation Day 10 was associated with a reduced pregnancy rate resulting from effects on sperm production in the F1-generation.
Lactation
Risk Summary
There are no data on the presence of cedazuridine, decitabine, or their metabolites in human milk or on their effects on the breastfed child or milk production. Because of the potential for serious adverse reactions in the breastfed child, advise women not to breastfeed during treatment with INQOVI and for 2 weeks after the last dose.
Females And Males Of Reproductive Potential
INQOVI can cause fetal harm when administered to a pregnant woman [see Use In Specific Populations].
Pregnancy Testing
Verify the pregnancy status in females of reproductive potential prior to initiating INQOVI.
Contraception
Females
Advise females of reproductive potential to use effective contraception during treatment with INQOVI and for 6 months after the last dose.
Males
Based on genotoxicity findings, advise males with female partners of reproductive potential to use effective contraception during treatment with INQOVI and for 3 months after the last dose [see Nonclinical Toxicology].
Infertility
Based on findings of decitabine and cedazuridine in animals, INQOVI may impair male fertility [see Nonclinical Toxicology]. The reversibility of the effect on fertility is unknown.
Pediatric Use
The safety and effectiveness of INQOVI have not been established in pediatric patients.
Geriatric Use
Of the 208 patients in clinical studies who received INQOVI, 75% were age 65 years and older, while 36% were age 75 years and older. No overall differences in safety or effectiveness were observed between patients age 65 years and older, 75 years and older, and younger patients.
Renal Impairment
No dosage modification of INQOVI is recommended for patients with mild or moderate renal impairment (creatinine clearance [CLcr] of 30 to 89 mL/min based on Cockcroft-Gault). Due to the potential for increased adverse reactions, monitor patients with moderate renal impairment (CLcr 30 to 59 mL/min) frequently for adverse reactions. INQOVI has not been studied in patients with severe renal impairment (CLcr 15 to 29 mL/min) or end-stage renal disease (ESRD: CLcr <15 mL/min) [see CLINICAL PHARMACOLOGY].
Clinical Pharmacology for Inqovi
Mechanism Of Action
Decitabine is a nucleoside metabolic inhibitor that is believed to exert its effects after phosphorylation and direct incorporation into DNA and inhibition of DNA methyltransferase, causing hypomethylation of DNA and cellular differentiation and/or apoptosis. Decitabine inhibits DNA methylation in vitro, which is achieved at concentrations that do not cause major suppression of DNA synthesis. Decitabine-induced hypomethylation in cancer cells may restore normal function to genes that are critical for the control of cellular differentiation and proliferation. In rapidly dividing cells, the cytotoxicity of decitabine may also be attributed to the formation of covalent adducts between DNA methyltransferase and decitabine incorporated into DNA. Non-proliferating cells are relatively insensitive to decitabine.
Cytidine deaminase (CDA) is an enzyme that catalyzes the degradation of cytidine, including the cytidine analog decitabine. High levels of CDA in the gastrointestinal tract and liver degrade decitabine and limit its oral bioavailability. Cedazuridine is a CDA inhibitor. Administration of cedazuridine with decitabine increases systemic exposure of decitabine.
Pharmacodynamics
Decitabine induced hypomethylation both in vitro and in vivo. In patients administered the recommended dosage of INQOVI, the maximum change from baseline in the long interspersed nucleotide elements-1 (LINE-1) demethylation was observed at Day 8, with less than complete recovery of LINE-1 methylation to baseline at the end of the treatment cycle.
Based on the exposure-response analyses, a relationship between an increase in 5-day cumulative daily decitabine exposure and a greater likelihood of some adverse reactions (e.g., any grade neutropenias, thrombocytopenia) was observed in clinical studies.
Pharmacokinetics
The pharmacokinetics of decitabine and cedazuridine following administration of INQOVI at the recommended dosage in patients with MDS and CMML are shown in Table 4.
The geometric mean ratio (GMR) of decitabine area under the curve (AUC) following the first dose of INQOVI compared to that of intravenous decitabine on Day 1 was 60% (90% confidence intervals (CI): 55, 65) in patients with MDS and CMML [see DOSAGE AND ADMINISTRATION]. The GMR of decitabine AUC following 5 consecutive once daily doses of INQOVI compared to that of intravenous decitabine on Day 5 was 106% (90% CI: 98, 114) and the GMR of the 5-day cumulative decitabine AUC following 5 consecutive once daily doses of INQOVI compared to that of intravenous decitabine was 99% (90% CI: 93, 106).
An approximately dose-proportional increase in peak concentrations (Cmax) and AUC over the dosing interval was observed for decitabine following administration of oral decitabine at 20 mg to 40 mg once daily (0.6 to 1.1 times the recommended dose) in combination with 100 mg oral cedazuridine, and for cedazuridine following administration of oral cedazuridine at 40 to 100 mg once daily (0.4 to 1.0 times the recommended dose) in combination with 20 mg oral decitabine.
Table 4: Pharmacokinetics of the Components of INQOVI*
| Parameter | Decitabine | Cedazuridine |
| General Information | ||
| With the recommended dosage of INQOVI for 5 consecutive days: | ||
| 5-day cumulative AUC, ng•hr/mL | 851 (50%) | -- |
| Day 1 AUC, ng•hr/mL | 103 (55%) | 2950 (49%) |
| Steady state AUC, ng•hr/mL | 178 (53%) | 3291 (45%) |
| Time to steady state, days | 2 | 2 |
| Accumulation ratio based on AUC | 1.7 (42%) | 1.1 (63%) |
| Cmax, ng/mL | 145 (55%) | 371 (52%) |
| Absorption | ||
| Bioavailability | Cedazuridine increases oral decitabine exposure | 20% (23%) |
| Tmax, hours‡ | 1 (0.3 to 3.0) | 3 (1.5 to 6.1) |
| Distribution | ||
| V/F at steady state, L | 417 (54%) | 296 (51%) |
| Fraction unbound, in vitro | 96% (4%) to 94% (2%) between 17 ng/mL to 342 ng/mL | 66% (6%) to 62% (2%) between 1000 ng/mL and 50000 ng/mL |
| Elimination | ||
| Half-life at steady state†, hours | 1.5 (27%) | 6.7 (19%) |
| CL/F at steady state, L/hours | 197 (53%) | 30.3 (46%) |
| Metabolism | ||
| Primary Pathways | Primarily by cytidine deaminase (CDA) and by physicochemical degradation | Conversion to epimer by physicochemical degradation |
| Excretion§ | ||
| Total (% unchanged) | -- | 46% (21%) in urine and 51% (27%) in feces |
| Cmax= maximum plasma concentration; AUC0-24h=area under the plasma concentration-time curve from time zero to 24 hours; CV=coefficient of variation; SD=standard deviation; Tmax= Time to maximum concentration; V/F=apparent volume of distribution; CL/F=apparent clearance * Mean (%CV) † Mean (SD) ‡ Median (range) § Healthy subjects |
||
Specific Populations
Age (32 to 90 years), sex, and mild hepatic impairment (total bilirubin > 1 to 1.5 × ULN or AST > ULN) did not have an effect on the pharmacokinetics of decitabine or cedazuridine after dosing with INQOVI.
Decitabine exposure (AUC) increased with decreasing body surface area or body weight, and cedazuridine exposure increased with decreasing CLcr; however, body surface area (1.3 to 2.9 m²), body weight (41 to 158 kg), and mild to moderate renal impairment (CLcr 30 to 89 mL/min based on Cockcroft Gault) did not have a clinically meaningful effect on the pharmacokinetics of decitabine and cedazuridine after dosing with INQOVI.
The effects of moderate (total bilirubin > 1.5 to 3 × ULN and any AST) and severe hepatic impairment (total bilirubin > 3 × ULN and any AST) or severe renal impairment (CLcr 15 to <30 mL/min) and ESRD (CLcr <15 mL/min) on the pharmacokinetics of decitabine and cedazuridine are unknown.
Drug Interaction Studies
Clinical Studies
Decitabine had no clinically meaningful effect on the pharmacokinetics of cedazuridine. Cedazuridine increased the exposure of decitabine.
The coadministration of INQOVI with proton pump inhibitors had no clinically meaningful effect on exposure to decitabine or cedazuridine.
In vitro Studies
CYP Enzymes: Cedazuridine is not a substrate of cytochrome P450 (CYP) enzymes. Cedazuridine does not induce CYP1A, CYP2B6, CYP2C9, or CYP3A or inhibit CYP1A, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, or CYP3A.
Transporter Systems: Cedazuridine is not a substrate of P-glycoprotein (P-gp), MATE1, MATE2-K, OAT1, OAT3, OATP1B1, OAPT1B3, OATP2B1, OCT1, or OCT2, and does not inhibit P-gp, BCRP, MATE1, MATE2-K, OAT1, OAT3, OATP1B1, OATP1B3, or OCT2.
Clinical Studies
Study ASTX727-01-B
INQOVI was evaluated in Study ASTX727-01-B, an open-label, randomized, 2-cycle, 2-sequence crossover study (NCT02103478) that included 80 adult patients with MDS (International Prognostic Scoring System [IPSS] Intermediate-1, Intermediate-2, or high-risk) or CMML. Patients were randomized 1:1 to receive INQOVI (35 mg decitabine and 100 mg cedazuridine) orally in Cycle 1 and decitabine 20 mg/m² intravenously in Cycle 2 or the reverse sequence. Both INQOVI and intravenous decitabine were administered once daily on Days 1 through 5 of the 28-day cycle. Starting with Cycle 3, all patients received INQOVI orally once daily on Days 1 through 5 of each 28-day cycle until disease progression or unacceptable toxicity. Randomization was stratified by IPSS risk level. Twelve (15%) of the 80 patients went on to stem cell transplantation following INQOVI treatment.
The baseline demographic and disease characteristics are shown in Table 5.
Table 5: Demographics and Baseline Disease Characteristics for Study ASTX727-01-B
| Characteristic | N=80 |
| Age | |
| Median (min, max) (years) | 71 (32, 90) |
| Sex (%) | |
| Male | 76 |
| Female | 24 |
| Race (%) | |
| White | 93 |
| Black or African American | 3 |
| Asian | 1 |
| Other or Not Reported | 4 |
| ECOG Performance Score (%) | |
| 0 | 44 |
| 1 | 48 |
| 2 | 9 |
| Disease Category / IPSS (%) | |
| MDS INT-1 | 44 |
| MDS INT-2 | 24 |
| MDS High-Risk | 11 |
| CMML | 21 |
| Prior HMA Therapy* (%) | |
| Prior Azacitidine | 4 |
| Prior Decitabine | 4 |
| Transfusion Dependence† (%) | |
| RBC Transfusion Dependence | 48 |
| Platelet Transfusion Dependence | 15 |
| * One cycle only, per the Exclusion Criteria. † Defined as documentation of ≥ 2 units of transfusion within 56 days prior to the first day of study treatment. |
|
Efficacy was established on the basis of complete response (CR) and the rate of conversion from transfusion dependence to transfusion independence. Efficacy results are shown in Table 6. The median follow-up time was 24.0 months (range: 12.0 to 28.8 months) and median treatment duration was 6.6 months (range < 0.1 to 27.9).
Table 6: Efficacy Results in Patients with MDS or CMML from Study ASTX727-01-B
| Efficacy Endpoint | INQOVI N=80 |
| Complete Response (%) (95% CI) | 18 (10, 28) |
| Median Duration of CR - months (range)* | 8.7 (1.1, 18.2) |
| Median Time to CR - months (range) | 4.8 (1.7, 10.0) |
| * From start of CR until relapse or death. | |
Among the 41 patients who were dependent on red blood cell (RBC) and/or platelet transfusions at baseline, 20 (49%) became independent of RBC and platelet transfusions during any consecutive 56-day post-baseline period. Of the 39 patients who were independent of both RBC and platelet transfusions at baseline, 25 (64%) remained transfusion-independent during any consecutive 56-day post-baseline period.
Study ASTX727-02
INQOVI was evaluated in ASTX727-02, an open-label, randomized, 2-cycle, 2-sequence crossover study (NCT03306264) that included 133 adult patients with MDS or CMML, including all French-American- British (FAB) classification criteria and IPSS Intermediate-1, Intermediate-2, or high-risk prognostic scores. Patients were randomized 1:1 to receive INQOVI (35 mg decitabine and 100 mg cedazuridine) orally in Cycle 1 and decitabine 20 mg/m² intravenously in Cycle 2 or the reverse sequence. Both INQOVI and intravenous decitabine were administered once daily on Days 1 through 5 of the 28-day cycle. Starting with Cycle 3, all patients received INQOVI orally once daily on Days 1 through 5 of each 28-day cycle until disease progression or unacceptable toxicity. No stratification was performed. Twentyseven (20%) of the 133 patients went on to stem cell transplantation following INQOVI treatment.
The baseline demographic and disease characteristics are shown in Table 7.
Table 7: Demographics and Baseline Disease Characteristics for Study ASTX727-02
| Characteristic | N=133 |
| Age (years) | |
| Median (min, max) | 71 (44, 88) |
| Sex (%) | |
| Male | 65 |
| Female | 35 |
| Race (%) | |
| White | 91 |
| Black or African American | 3 |
| Asian | 2 |
| Other or Not Reported | 4 |
| ECOG Performance Score (%) | |
| 0 | 41 |
| 1 | 59 |
| Disease Category / IPSS (%) | |
| MDS INT-1 | 44 |
| MDS INT-2 | 20 |
| MDS High Risk | 16 |
| MDS Low Risk | 8 |
| CMML | 12 |
| Prior HMA Therapy* (%) | |
| Prior Azacitidine | 5 |
| Prior Decitabine | 3 |
| Transfusion Dependence† (%) | |
| RBC Transfusion Dependence | 39 |
| Platelet Transfusion Dependence | 8 |
| * One cycle only, per the Exclusion Criteria. † Defined as documentation of ≥ 2 units of transfusion within 56 days prior to the first day of study treatment. |
|
The primary outcome measure was comparison of the 5-day cumulative decitabine AUC between INQOVI and intravenous decitabine [see CLINICAL PHARMACOLOGY]. Efficacy was established on the basis of complete response (CR) and the rate of conversion from transfusion dependence to transfusion independence. Efficacy results are shown in Table 8. The median follow-up time was 12.6 months (range: 9.3 to 20.5) and median treatment duration was 8.2 months (range 0.2 to 19.7).
Table 8: Efficacy Results in Patients with MDS or CMML from Study ASTX727-02
| Efficacy Endpoints | INQOVI (N=133) |
| Complete Response (%) (95% CI) | 21(15, 29) |
| Median Duration of CR - months (range)* | 7.5 (1.6, 17.5) |
| Median Time to CR - months (range) | 4.3 (2.1, 15.2) |
| * From start of CR until relapse or death. | |
Among the 57 patients who were dependent on RBC and/or platelet transfusions at baseline, 30 (53%) became independent of RBC and platelet transfusions during any 56-day post-baseline period. Of the 76 patients who were independent of both RBC and platelet transfusions at baseline, 48 (63%) remained transfusion-independent during any 56-day post-baseline period.
From 
Report Problems to the Food and Drug Administration
You are encouraged to report negative side effects of prescription drugs to the FDA. Visit the FDA MedWatch website or call 1-800-FDA-1088.