Description for Jakafi
Ruxolitinib phosphate is a kinase inhibitor with the chemical name (R)-3-(4-(7H-pyrrolo[2,3d] pyrimidin-4-yl)-1H-pyrazol-1-yl)-3-cyclopentylpropanenitrile phosphate and a molecular weight of 404.36. Ruxolitinib phosphate has the following structural formula:
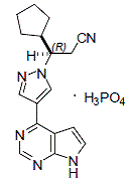 |
Ruxolitinib phosphate is a white to off-white to light pink powder and is soluble in aqueous buffers across a pH range of 1 to 8.
Jakafi (ruxolitinib) Tablets are for oral administration. Each tablet contains ruxolitinib phosphate equivalent to 5 mg, 10 mg, 15 mg, 20 mg and 25 mg of ruxolitinib free base together with microcrystalline cellulose, lactose monohydrate, magnesium stearate, colloidal silicon dioxide, sodium starch glycolate, povidone and hydroxypropyl cellulose.
Uses for Jakafi
Myelofibrosis
Jakafi is indicated for treatment of intermediate or high-risk myelofibrosis (MF), including primary MF, post-polycythemia vera MF and post-essential thrombocythemia MF in adults.
Polycythemia Vera
Jakafi is indicated for treatment of polycythemia vera (PV) in adults who have had an inadequate response to or are intolerant of hydroxyurea.
Acute Graft-Versus-Host Disease
Jakafi is indicated for treatment of steroid-refractory acute graft-versus-host disease (aGVHD) in adult and pediatric patients 12 years and older.
Chronic Graft-Versus-Host Disease
Jakafi is indicated for treatment of chronic graft-versus-host disease (cGVHD) after failure of one or two lines of systemic therapy in adult and pediatric patients 12 years and older.
Dosage for Jakafi
Monitoring To Assess Safety
Prior To Jakafi Treatment
- Perform a complete blood count [see WARNINGS AND PRECAUTIONS].
- Inquire about past infections, including tuberculosis, herpes simplex, herpes zoster, and hepatitis B [see WARNINGS AND PRECAUTIONS].
During Treatment With Jakafi
- Perform a complete blood count every 2 to 4 weeks until doses are stabilized, and then as clinically indicated [see WARNINGS AND PRECAUTIONS].
- Assess lipid parameters approximately 8-12 weeks following initiation of Jakafi therapy [see WARNINGS AND PRECAUTIONS].
Recommended Dosage For Myelofibrosis
The recommended starting dose of Jakafi is based on platelet count (Table 1). Doses may be titrated based on safety and efficacy.
Table 1: Jakafi Starting Doses for Myelofibrosis
| Platelet Count | Starting Dose |
| Greater than 200 x 109/L | 20 mg orally twice daily |
| 100 x 109/L to 200 x 109/L | 15 mg orally twice daily |
| 50 x 109/L to less than 100 x 109/L | 5 mg orally twice daily |
Dose Modification Guidelines Fir Hematologic Toxicity For Patients With Myelofibrosis Starting Treatment With A Platelet Count Of 100 × 109/L Or Greater
Treatment Interruption And Restarting Dosing
Interrupt treatment for platelet counts less than 50 × 109/L or absolute neutrophil count (ANC) less than 0.5 × 109/L.
After recovery of platelet counts above 50 × 109/L and ANC above 0.75 × 109/L, dosing may be restarted. Table 2 illustrates the maximum allowable dose that may be used in restarting Jakafi after a previous interruption.
Table 2: Myelofibrosis: Maximum Restarting Doses for Jakafi after Safety Interruption for Thrombocytopenia for Patients Starting Treatment with a Platelet Count of 100 × 109/L or Greater
| Current Platelet Count | Maximum Dose When Restarting Jakafi Treatment* |
| Greater than or equal to 125 x 109/L | 20 mg twice daily |
| 100 to less than 125 x 109/L | 15 mg twice daily |
| 75 to less than 100 x 109/L | 10 mg twice daily for at least 2 weeks; if stable, may increase to 15 mg twice daily |
| 50 to less than 75 x 109/L | 5 mg twice daily for at least 2 weeks; if stable, may increase to 10 mg twice daily |
| Less than 50 x 109/L | Continue hold |
| *Maximum doses are displayed. When restarting, begin with a dose at least 5 mg twice daily below the dose at interruption. | |
Following treatment interruption for ANC below 0.5 × 109/L, after ANC recovers to 0.75 × 109/L or greater, restart dosing at the higher of 5 mg once daily or 5 mg twice daily below the largest dose in the week prior to the treatment interruption.
Dose Reductions
Dose reductions should be considered if the platelet counts decrease as outlined in Table 3 with the goal of avoiding dose interruptions for thrombocytopenia.
Table 3: Myelofibrosis: Dosing Recommendations for Thrombocytopenia for Patients Starting Treatment with a Platelet Count of 100 × 109/L or Greater
| Platelet Count | Dose at Time of Platelet Decline | ||||
| 25 mg twice daily | 20 mg twice daily | 15 mg twice daily | 10 mg twice daily | 5 mg twice daily | |
| New Dose | New Dose | New Dose | New Dose | New Dose | |
| 100 to less than 125 x 109/L | 20 mg twice daily | 15 mg twice daily | No Change | No Change | No Change |
| 75 to less than 100 x 109/L | 10 mg twice daily | 10 mg twice daily | 10 mg twice daily | No Change | No Change |
| 50 to less than 75 x 109/L | 5 mg twice daily | 5 mg twice daily | 5 mg twice daily | 5 mg twice daily | No Change |
| Less than 50 x 109/L | Hold | Hold | Hold | Hold | Hold |
Dose Modification Based On Insufficient Response For Patients With Myelofibrosis Starting Treatment With A Platelet Count Of 100 × 109/L or Greater
If the response is insufficient and platelet and neutrophil counts are adequate, doses may be increased in 5 mg twice daily increments to a maximum of 25 mg twice daily. Doses should not be increased during the first 4 weeks of therapy and not more frequently than every 2 weeks.
Consider dose increases in patients who meet all of the following conditions:
- Failure to achieve a reduction from pretreatment baseline in either palpable spleen length of 50% or a 35% reduction in spleen volume as measured by computed tomography (CT) or magnetic resonance imaging (MRI);
- Platelet count greater than 125 × 109/L at 4 weeks and platelet count never below 100 × 109/L;
- ANC Levels greater than 0.75 × 109/L.
Based on limited clinical data, long-term maintenance at a 5 mg twice daily dose has not shown responses and continued use at this dose should be limited to patients in whom the benefits outweigh the potential risks. Discontinue Jakafi if there is no spleen size reduction or symptom improvement after 6 months of therapy.
Dose Modifications For Hematologic Toxicity For Patients With Myelofibrosis Starting Treatment With Platelet Counts Of 50 × 109/L To Less Than 100 × 109/L
This section applies only to patients with platelet counts of 50 × 109/L to less than 100 × 109/L prior to any treatment with Jakafi. See dose modifications in Section 2.2 (Dose Modification Guidelines for Hematological Toxicity for Patients with Myelofibrosis Starting Treatment with a Platelet Count of 100 × 109/L or Greater) for hematological toxicity in patients whose platelet counts were 100 × 109/L or more prior to starting treatment with Jakafi.
Treatment Interruption And Restarting Dosing
Interrupt treatment for platelet counts less than 25 × 109/L or ANC less than 0.5 × 109/L.
After recovery of platelet counts above 35 × 109/L and ANC above 0.75 × 109/L, dosing may be restarted. Restart dosing at the higher of 5 mg once daily or 5 mg twice daily below the largest dose in the week prior to the decrease in platelet count below 25 × 109/L or ANC below 0.5 × 109/L that led to dose interruption.
Dose Reductions
Reduce the dose of Jakafi for platelet counts less than 35 × 109/L as described in Table 4.
Table 4: Myelofibrosis: Dosing Modifications for Thrombocytopenia for Patients with Starting Platelet Count of 50 × 109/L to Less Than 100 × 109/L
| Platelet Count | Dosing Recommendations |
| Less than 25 x 109/L |
|
| 25 x 109/L to less than 35 x 109/L AND the platelet count decline is less than 20% during the prior four weeks |
|
| 25 x 109/L to less than 35 x 109/L AND the platelet count decline is 20% or greater during the prior four weeks |
|
Dose Modifications Based On Insufficient Response For Patients With Myelofibrosis And Starting Platelet Count Of 50 × 109/L To Less Than 100 × 109/L
Do not increase doses during the first 4 weeks of therapy, and do not increase the dose more frequently than every 2 weeks.
If the response is insufficient as defined in Section 2.2 (see Dose Modification Based on Insufficient Response with Myelofibrosis Starting Treatment with a platelet count of 100 × 109/L or Greater), doses may be increased by increments of 5 mg daily to a maximum of 10 mg twice daily if:
- the platelet count has remained at least 40 × 109/L, and
- the platelet count has not fallen by more than 20% in the prior 4 weeks, and
- the ANC is more than 1 × 109/L, and
- the dose has not been reduced or interrupted for an adverse event or hematological toxicity in the prior 4 weeks.
Continuation of treatment for more than 6 months should be limited to patients in whom the benefits outweigh the potential risks. Discontinue Jakafi if there is no spleen size reduction or symptom improvement after 6 months of therapy.
Dose Modification For Bleeding
Interrupt treatment for bleeding requiring intervention regardless of current platelet count. Once the bleeding event has resolved, consider resuming treatment at the prior dose if the underlying cause of bleeding has been controlled. If the bleeding event has resolved but the underlying cause persists, consider resuming treatment with Jakafi at a lower dose.
Recommended Dosage For Polycythemia Vera
The recommended starting dose of Jakafi is 10 mg twice daily. Doses may be titrated based on safety and efficacy.
Dose Modification Guidelines For Patients With Polycythemia Vera
Dose Reductions
Dose reductions should be considered for hemoglobin and platelet count decreases as described in Table 5.
Table 5: Polycythemia Vera: Dose Reductions
| Hemoglobin and/or Platelet Count | Dosing Recommendations |
| Hemoglobin greater than or equal to 12 g/dL AND platelet count greater than or equal to 100 x 109/L |
|
| Hemoglobin 10 to less than 12 g/dL AND platelet count 75 to less than 100 x 109/L |
|
| Hemoglobin 8 to less than 10 g/dL OR platelet count 50 to less than 75 x 109/L |
|
| Hemoglobin less than 8 g/dL OR platelet count less than 50 x 109/L |
|
Treatment Interruption And Restarting Dosing
Interrupt treatment for hemoglobin less than 8 g/dL, platelet counts less than 50 × 109/L or ANC less than 1.0 × 109/L.
After recovery of the hematologic parameter(s) to acceptable levels, dosing may be restarted.
Table 6 illustrates the dose that may be used in restarting Jakafi after a previous interruption.
Table 6: Polycythemia Vera: Restarting Doses for Jakafi after Safety Interruption for Hematologic Parameter(s)
Use the most severe category of a patient’s hemoglobin, platelet count, or ANC abnormality to determine the corresponding maximum restarting dose.
| Hemoglobin, Platelet Count, or ANC | Maximum Restarting Dose |
| Hemoglobin less than 8 g/dL OR platelet count less than 50 x 109/L OR ANC less than 1 x 109/L | Continue hold |
| Hemoglobin 8 to less than 10 g/dL OR platelet count 50 to less than 75 x 109/L OR ANC 1 to less than 1.5 x 109/L | 5 mg twice dailya or no more than 5 mg twice daily less than the dose which resulted in dose interruption |
| Hemoglobin 10 to less than 12 g/dL OR platelet count 75 to less than 100 x 109/L OR ANC 1.5 to less than 2 x 109/L | 10 mg twice dailya or no more than 5 mg twice daily less than the dose which resulted in dose interruption |
| Hemoglobin greater than or equal to 12 g/dL OR platelet count greater than or equal to 100 x 109/L OR ANC greater than or equal to 2 x 109/L | 15 mg twice dailya or no more than 5 mg twice daily less than the dose which resulted in dose interruption |
| a Continue treatment for at least 2 weeks; if stable, may increase dose by 5 mg twice daily. | |
Patients who had required dose interruption while receiving a dose of 5 mg twice daily, may restart at a dose of 5 mg twice daily or 5 mg once daily, but not higher, once hemoglobin is greater than or equal to 10 g/dL, platelet count is greater than or equal to 75 × 109/L, and ANC is greater than or equal to 1.5 × 109/L.
Dose Management After Restarting Treatment
After restarting Jakafi following treatment interruption, doses may be titrated, but the maximum total daily dose should not exceed 5 mg less than the dose that resulted in the dose interruption. An exception to this is dose interruption following phlebotomy-associated anemia, in which case the maximal total daily dose allowed after restarting Jakafi would not be limited.
Dose Modifications Based On Insufficient Response For Patients With Polycythemia Vera
If the response is insufficient and platelet, hemoglobin, and neutrophil counts are adequate, doses may be increased in 5 mg twice daily increments to a maximum of 25 mg twice daily. Doses should not be increased during the first 4 weeks of therapy and not more frequently than every two weeks.
Consider dose increases in patients who meet all of the following conditions:
- Inadequate efficacy as demonstrated by one or more of the following:
- Continued need for phlebotomy
- WBC greater than the upper limit of normal range
- Platelet count greater than the upper limit of normal range
- Palpable spleen that is reduced by less than 25% from Baseline
- Platelet count greater than or equal to 140 × 109/L
- Hemoglobin greater than or equal to 12 g/dL
- ANC greater than or equal to 1.5 × 109/L
Recommended Dosage For Acute Graft-Versus-Host Disease
The recommended starting dose of Jakafi is 5 mg given orally twice daily. Consider increasing the dose to 10 mg twice daily after at least 3 days of treatment if the ANC and platelet counts are not decreased by 50% or more relative to the first day of dosing with Jakafi.
Consider tapering Jakafi after 6 months of treatment in patients with response who have discontinued therapeutic doses of corticosteroids. Taper Jakafi by one dose level approximately every 8 weeks (10 mg twice daily to 5 mg twice daily to 5 mg once daily). If aGVHD signs or symptoms recur during or after the taper of Jakafi, consider retreatment.
Dose Modification Guidelines For Patients With Acute Graft-Versus-Host Disease
Monitor complete blood counts (CBC), including platelet count and ANC, and bilirubin prior to initiating therapy, every 2 to 4 weeks until doses are stabilized, and then as indicated clinically.
Modify the dose of Jakafi for adverse reactions as described in Table 7. For dose reductions, patients who are currently receiving Jakafi 10 mg twice daily may have their dose reduced to 5 mg twice daily; patients receiving 5 mg twice daily may have their dose reduced to 5 mg once daily. Patients who are unable to tolerate Jakafi at a dose of 5 mg once daily should have treatment interrupted until their clinical and/or laboratory parameters recover.
Table 7: Dose Modifications for Adverse Reactions in Patients with Acute GVHD
| Laboratory Parameter | Dosing Recommendations |
| Clinically significant thrombocytopenia after supportive measures | Reduce dose by 1 dose level. When platelets recover to previous values, dosing may return to prior dose level. |
| ANC less than 1 x 109/L considered related to Jakafi | Hold Jakafi for up to 14 days; resume at 1 dose level lower upon recovery. |
| Total Bilirubin elevation, no liver GVHD | 3.0-5.0 x ULN: Continue Jakafi at 1 dose level lower until recovery. > 5.0-10.0 x ULN: Hold Jakafi for up to 14 days until bilirubin ≤ 1.5 x ULN; resume at current dose upon recovery. Total bilirubin > 10.0 x ULN: Hold Jakafi for up to 14 days until bilirubin ≤ 1.5 x ULN; resume at 1 dose level lower upon recovery. |
| Total Bilirubin elevation, liver GVHD | > 3.0 x ULN: Continue Jakafi at 1 dose level lower until recovery. |
Recommended Dosage For Chronic Graft-Versus-Host Disease
The recommended starting dose of Jakafi is 10 mg given orally twice daily.
Consider tapering Jakafi after 6 months of treatment in patients with response who have discontinued therapeutic doses of corticosteroids. Taper Jakafi by one dose level approximately every 8 weeks (10 mg twice daily to 5 mg twice daily to 5 mg once daily). If GVHD signs or symptoms recur during or after the taper of Jakafi, consider retreatment.
Dose Modification Guidelines For Patients With Chronic Graft-Versus-Host Disease
Monitor complete blood counts (CBC), including platelet count and ANC, and bilirubin prior to initiating therapy, every 2 to 4 weeks until doses are stabilized, and then as indicated clinically.
Modify the dose of Jakafi for adverse reactions as described in Table 8. For dose reductions, patients who are currently receiving Jakafi 10 mg twice daily may have their dose reduced to 5 mg twice daily; patients receiving 5 mg twice daily may have their dose reduced to 5 mg once daily. Patients who are unable to tolerate Jakafi at a dose of 5 mg once daily should have treatment interrupted until their clinical and/or laboratory parameters recover.
Table 8: Dose Modifications for Adverse Reactions in Patients with Chronic GVHD
| Parameter | Dosing Recommendations |
| Platelet count less than 20 x 109/L | Reduce Jakafi by 1 dose level. If resolved within 7 days, dosing may return to initial dose level. If not resolved within 7 days, then maintain at 1 dose level lower. |
| ANC less than 0.75 x 109/L considered related to Jakafi | Reduce Jakafi by 1 dose level; resume at initial dose level upon recovery. |
| ANC less than 0.5 x 109/L considered related to Jakafi | Hold Jakafi for up to 14 days; resume at 1 dose level lower upon recovery. May resume initial dose level when ANC greater than 1.0 x 109/L. |
| Total Bilirubin: 3.0-5.0 x ULN | Continue Jakafi at 1 dose level lower until recovery. If resolved within 14 days, then increase by one dose level. If not resolved within 14 days, then maintain the decreased dose level. |
| Total Bilirubin: > 5.0-10.0 x ULN | Hold Jakafi for up to 14 days until resolved; resume at current dose upon recovery. If not resolved within 14 days, then resume at 1 dose level lower upon recovery. |
| Total Bilirubin: > 10.0 x ULN | Hold Jakafi for up to 14 days until resolved; resume at 1 dose level lower upon recovery. If not resolved within 14 days, discontinue. |
| Other Adverse Reactions: Grade 3 | Continue Jakafi at 1 dose level lower until recovery. |
| Other Adverse Reactions: Grade 4 | Discontinue Jakafi. |
Dose Modifications For Concomitant Use With Strong CYP3A4 Inhibitors Or Fluconazole
Modify the Jakafi dosage when coadministered with strong CYP3A4 inhibitors or doses of less than or equal to 200 mg of fluconazole [see DRUG INTERACTIONS], according to Table 9. Avoid concomitant use of Jakafi with fluconazole doses of greater than 200 mg daily.
Table 9: Dose Modifications for Concomitant Use with Strong CYP3A4 Inhibitors or Fluconazole
| For patients coadministered strong CYP3A4 inhibitors or doses of less than or equal to 200 mg of fluconazole | Recommended Jakafi Dose Modification |
| Starting dose for patients with MF with a platelet count: | |
|
10 mg twice daily |
|
5 mg once daily |
| Starting dose for patients with PV: | 5 mg twice daily |
| If on stable dose for patients with MF or PV: | |
|
Decrease dose by 50% (round up to the closest available tablet strength) |
|
5 mg once daily |
|
Avoid strong CYP3A4 inhibitor or fluconazole treatment or interrupt Jakafi treatment for the duration of strong CYP3A4 inhibitor or fluconazole use |
| Starting dose for patients with aGVHD or cGVHD: | |
| Fluconazole doses of less than or equal to 200 mg | 5 mg once daily for patients with aGVHD; 5 mg twice daily for patients with cGVHD |
| Other CYP3A4 inhibitors | Monitor blood counts more frequently for toxicity and modify the Jakafi dosage for adverse reactions if they occur [see DOSAGE AND ADMINISTRATION]. |
Dose Modifications For Renal Or Hepatic Impairment
Moderate To Severe Renal Impairment Or End Stage Renal Disease On Dialysis
Modify the Jakafi dosage for patients with moderate (CLcr 30 to 59 mL/min) to severe (CLcr 15 to 29 mL/min) renal impairment or end stage renal disease (ESRD) on dialysis according to
Table 10: Avoid use of Jakafi in patients with ESRD (CLcr less than 15 mL/min) not requiring dialysis [see Use In Specific Populations].
Table 10: Dose Modifications for Renal Impairment
| Renal Impairment Status | Platelet Count | Recommended Starting Dosage |
| Patients with MF | ||
| Moderate or Severe | Greater than 150 x 109/L | No dose adjustment |
| 100 to 150 x 109/L | 10 mg twice daily | |
| 50 to less than 100 x 109/L | 5 mg daily | |
| Less than 50 x 109/L | Avoid use [see Use In Specific Populations] | |
| ESRD on dialysis | 100 to 200 x 109/L | 15 mg once after dialysis session |
| Greater than 200 x 109/L | 20 mg once after dialysis session | |
| Patients with PV | ||
| Moderate or Severe | Any | 5 mg twice daily |
| ESRD on dialysis | Any | 10 mg once after dialysis session |
| Patients with aGVHD | ||
| Moderate or Severe | Any | 5 mg once daily |
| ESRD on dialysis | Any | 5 mg once after dialysis session |
| Patients with cGVHD | ||
| Moderate or Severe | Any | 5 mg twice daily |
| ESRD on dialysis | Any | 10 mg once after dialysis session |
| ESRD = end stage renal disease and CLcr = creatinine clearance | ||
Hepatic Impairment
Modify the Jakafi dosage for patients with hepatic impairment according to Table 11.
Table 11: Dose Modifications for Hepatic Impairment
| Hepatic Impairment Status | Platelet Count | Recommended Starting Dosage |
| Patients with MF Mild, Moderate, or Severe (Child-Pugh Class A, B, C) | Greater than 150 x 109/L | No dose adjustment |
| 100 x 109/L to 150 x 109/L | 10 mg twice daily | |
| 50 to less than 100 x 109/L | 5 mg daily | |
| Less than 50 x 109/L | Avoid use [see Use In Specific Populations] | |
| Patients with PV Mild, Moderate, or Severe (Child-Pugh Class A, B, C) | Any | 5 mg twice daily |
| Patients with aGVHD | ||
| Mild, Moderate, or Severe based on NCI criteria without liver GVHD | Any | No dose adjustment |
| Stage 1, 2 or 3 Liver aGVHD | Any | No dose adjustment |
| Stage 4 Liver aGVHD | Any | 5 mg once daily |
| Patients with cGVHD | ||
| Mild, Moderate, or Severe based on NCI criteria without liver GVHD | Any | No dose adjustment |
| Score 1 or 2 Liver cGVHD | Any | No dose adjustment |
| Score 3 Liver cGVHD | Any | Monitor blood counts more frequently for toxicity and modify the Jakafi dosage for adverse reactions if they occur [see DOSAGE AND ADMINISTRATION]. |
Method Of Administration
Jakafi is dosed orally and can be administered with or without food.
If a dose is missed, the patient should not take an additional dose, but should take the next usual prescribed dose.
When discontinuing Jakafi therapy for reasons other than thrombocytopenia, gradual tapering of the dose of Jakafi may be considered, for example by 5 mg twice daily each week.
For patients unable to ingest tablets, Jakafi can be administered through a nasogastric tube (8 French or greater) as follows:
- Suspend one tablet in approximately 40 mL of water with stirring for approximately 10 minutes.
- Within 6 hours after the tablet has dispersed, the suspension can be administered through a nasogastric tube using an appropriate syringe.
The tube should be rinsed with approximately 75 mL of water. The effect of tube feeding preparations on Jakafi exposure during administration through a nasogastric tube has not been evaluated.
HOW SUPPLIED
Dosage Forms And Strengths
5 mg tablets -round and white with “INCY” on one side and “5” on the other.
10 mg tablets -round and white with “INCY” on one side and “10” on the other.
15 mg tablets -oval and white with “INCY” on one side and “15” on the other.
20 mg tablets -capsule-shaped and white with “INCY” on one side and “20” on the other.
25 mg tablets -oval and white with “INCY” on one side and “25” on the other.
Storage And Handling
Jakafi (ruxolitinib) Tablets are available as follows:
Jakafi Trade Presentations
| NDC Number | Strength | Description | Tablets per Bottle |
| 50881-005-60 | 5 mg | Round tablet with “INCY” on one side and “5” on the other | 60 |
| 50881-010-60 | 10 mg | Round tablet with “INCY” on one side and “10” on the other | 60 |
| 50881-015-60 | 15 mg | Oval tablet with “INCY” on one side and “15” on the other | 60 |
| 50881-020-60 | 20 mg | Capsule-shaped tablet with “INCY” on one side and “20” on the other | 60 |
| 50881-025-60 | 25 mg | Oval tablet with “INCY” on one side and “25” on the other | 60 |
Store at room temperature 20°C to 25°C (68°F to 77°F); excursions permitted between 15°C and 30°C (59°F and 86°F) [see USP Controlled Room Temperature].
Manufactured for: Incyte Corporation 1801 Augustine Cut-off Wilmington, DE 19803. Revised: Jan 2023
Side Effects for Jakafi
The following clinically significant adverse reactions are discussed in greater detail in other sections of the labeling:
- Thrombocytopenia, Anemia and Neutropenia [see WARNINGS AND PRECAUTIONS]
- Risk of Infection [see WARNINGS AND PRECAUTIONS]
- Symptom Exacerbation Following Interruption or Discontinuation of Treatment with Jakafi [see WARNINGS AND PRECAUTIONS]
- Non-Melanoma Skin Cancer [see WARNINGS AND PRECAUTIONS]
- Lipid Elevations [see WARNINGS AND PRECAUTIONS]
- Major Adverse Cardiovascular Events (MACE) [see WARNINGS AND PRECAUTIONS]
- Thrombosis [see WARNINGS AND PRECAUTIONS]
- Secondary Malignancies [see WARNINGS AND PRECAUTIONS]
Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Myelofibrosis
The safety of Jakafi was assessed in 617 patients in six clinical studies with a median duration of follow-up of 10.9 months, including 301 patients with MF in two Phase 3 studies.
In these two Phase 3 studies, patients had a median duration of exposure to Jakafi of 9.5 months (range 0.5 to 17 months), with 89% of patients treated for more than 6 months and 25% treated for more than 12 months. One hundred and eleven (111) patients started treatment at 15 mg twice daily and 190 patients started at 20 mg twice daily. In patients starting treatment with 15 mg twice daily (pretreatment platelet counts of 100 to 200 x 109/L) and 20 mg twice daily (pretreatment platelet counts greater than 200 x 109/L), 65% and 25% of patients, respectively, required a dose reduction below the starting dose within the first 8 weeks of therapy.
In a double-blind, randomized, placebo-controlled study of Jakafi, among the 155 patients treated with Jakafi, the most frequent adverse reactions were thrombocytopenia and anemia [see Table 13]. Thrombocytopenia, anemia and neutropenia are dose-related effects. The three most frequent nonhematologic adverse reactions were bruising, dizziness and headache [see Table 12].
Discontinuation for adverse events, regardless of causality, was observed in 11% of patients treated with Jakafi and 11% of patients treated with placebo.
Table 12 presents the most common nonhematologic adverse reactions occurring in patients who received Jakafi in the double-blind, placebo-controlled study during randomized treatment.
Table 12: Myelofibrosis: Nonhematologic Adverse Reactions Occurring in Patients on Jakafi in the Double-blind, Placebo-controlled Study During Randomized Treatment
| Adverse Reactions | Jakafi (N=155) |
Placebo (N=151) |
||||
| All Gradesa (%) | Grade 3 (%) | Grade 4 (%) | All Grades (%) | Grade 3 (%) | Grade 4 (%) | |
| Bruisingb | 23 | < 1 | 0 | 15 | 0 | 0 |
| Dizzinessc | 18 | < 1 | 0 | 7 | 0 | 0 |
| Headache | 15 | 0 | 0 | 5 | 0 | 0 |
| Urinary Tract Infectionsd | 9 | 0 | 0 | 5 | < 1 | < 1 |
| Weight Gaine | 7 | < 1 | 0 | 1 | < 1 | 0 |
| Flatulence | 5 | 0 | 0 | < 1 | 0 | 0 |
| Herpes Zosterf | 2 | 0 | 0 | < 1 | 0 | 0 |
| a National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE), version 3.0 b includes contusion, ecchymosis, hematoma, injection site hematoma, periorbital hematoma, vessel puncture site hematoma, increased tendency to bruise, petechiae, purpura c includes dizziness, postural dizziness, vertigo, balance disorder, Meniere's Disease, labyrinthitis d includes urinary tract infection, cystitis, urosepsis, urinary tract infection bacterial, kidney infection, pyuria, bacteria urine, bacteria urine identified, nitrite urine present e includes weight increased, abnormal weight gain f includes herpes zoster and post-herpetic neuralgia |
||||||
Description Of Selected Adverse Reactions
Anemia
In the two Phase 3 clinical studies, median time to onset of first CTCAE Grade 2 or higher anemia was approximately 6 weeks. One patient (< 1%) discontinued treatment because of anemia. In patients receiving Jakafi, mean decreases in hemoglobin reached a nadir of approximately 1.5 to 2.0 g/dL below baseline after 8 to 12 weeks of therapy and then gradually recovered to reach a new steady state that was approximately 1.0 g/dL below baseline. This pattern was observed in patients regardless of whether they had received transfusions during therapy.
In the randomized, placebo-controlled study, 60% of patients treated with Jakafi and 38% of patients receiving placebo received red blood cell transfusions during randomized treatment. Among transfused patients, the median number of units transfused per month was 1.2 in patients treated with Jakafi and 1.7 in placebo treated patients.
Thrombocytopenia
In the two Phase 3 clinical studies, in patients who developed Grade 3 or 4 thrombocytopenia, the median time to onset was approximately 8 weeks. Thrombocytopenia was generally reversible with dose reduction or dose interruption. The median time to recovery of platelet counts above 50 x 109/L was 14 days. Platelet transfusions were administered to 5% of patients receiving Jakafi and to 4% of patients receiving control regimens. Discontinuation of treatment because of thrombocytopenia occurred in < 1% of patients receiving Jakafi and < 1% of patients receiving control regimens. Patients with a platelet count of 100 x 109/L to 200 x 109/L before starting Jakafi had a higher frequency of Grade 3 or 4 thrombocytopenia compared to patients with a platelet count greater than 200 x 109/L (17% versus 7%).
Neutropenia
In the two Phase 3 clinical studies, 1% of patients reduced or stopped Jakafi because of neutropenia.
Table 13 provides the frequency and severity of clinical hematology abnormalities reported for patients receiving treatment with Jakafi or placebo in the placebo-controlled study.
Table 13: Myelofibrosis: Worst Hematology Laboratory Abnormalities in the Placebo-Controlled Studya
| Laboratory Parameter | Jakafi (N=155) |
Placebo (N=151) |
||||
| All Gradesb (%) | Grade 3 (%) | Grade 4 (%) | All Grades (%) | Grade 3 (%) | Grade 4 (%) | |
| Thrombocytopenia | 70 | 9 | 4 | 31 | 1 | 0 |
| Anemia | 96 | 34 | 11 | 87 | 16 | 3 |
| Neutropenia | 19 | 5 | 2 | 4 | < 1 | 1 |
| a Presented values are worst Grade values regardless of baseline b National Cancer Institute Common Terminology Criteria for Adverse Events, version 3.0 |
||||||
Additional Data From The Placebo-Controlled Study
- 25% of patients treated with Jakafi and 7% of patients treated with placebo developed newly occurring or worsening Grade 1 abnormalities in alanine transaminase (ALT). The incidence of greater than or equal to Grade 2 elevations was 2% for Jakafi with 1% Grade 3 and no Grade 4 ALT elevations.
- 17% of patients treated with Jakafi and 6% of patients treated with placebo developed newly occurring or worsening Grade 1 abnormalities in aspartate transaminase (AST). The incidence of Grade 2 AST elevations was < 1% for Jakafi with no Grade 3 or 4 AST elevations.
- 17% of patients treated with Jakafi and < 1% of patients treated with placebo developed newly occurring or worsening Grade 1 elevations in cholesterol. The incidence of Grade 2 cholesterol elevations was < 1% for Jakafi with no Grade 3 or 4 cholesterol elevations.
Polycythemia Vera
In a randomized, open-label, active-controlled study, 110 patients with PV resistant to or intolerant of hydroxyurea received Jakafi and 111 patients received best available therapy [see Clinical Studies]. The most frequent adverse reaction was anemia. Discontinuation for adverse events, regardless of causality, was observed in 4% of patients treated with Jakafi. Table 14 presents the most frequent nonhematologic adverse reactions occurring up to Week 32.
Table 14: Polycythemia Vera: Nonhematologic Adverse Reactions Occurring in ≥ 5% of Patients on Jakafi in the Open-Label, Active-controlled Study up to Week 32 of Randomized Treatment
| Adverse Reactions | Jakafi (N=110) |
Best Available Therapy (N=111) |
||
| All Gradesa (%) | Grade 3-4 (%) | All Grades (%) | Grade 3-4 (%) | |
| Diarrhea | 15 | 0 | 7 | < 1 |
| Dizzinessb | 15 | 0 | 13 | 0 |
| Dyspneac | 13 | 3 | 4 | 0 |
| Muscle Spasms | 12 | < 1 | 5 | 0 |
| Constipation | 8 | 0 | 3 | 0 |
| Herpes Zosterd | 6 | < 1 | 0 | 0 |
| Nausea | 6 | 0 | 4 | 0 |
| Weight Gaine | 6 | 0 | < 1 | 0 |
| Urinary Tract Infectionsf | 6 | 0 | 3 | 0 |
| Hypertension | 5 | < 1 | 3 | < 1 |
| a National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE), version 3.0 b includes dizziness and vertigo c includes dyspnea and dyspnea exertional d includes herpes zoster and post-herpetic neuralgia e includes weight increased and abnormal weight gain f includes urinary tract infection and cystitis |
||||
Clinically relevant laboratory abnormalities are shown in Table 15.
Table 15: Polycythemia Vera: Selected Laboratory Abnormalities in the Open-Label, Active-controlled Study up to Week 32 of Randomized Treatmenta
| Laboratory Parameter | Jakafi (N=110) |
Best Available Therapy (N=111) |
||||
| All Gradesb (%) | Grade 3m(%) | Grade 4 (%) | All Grades (%) | Grade 3 (%) | Grade 4 (%) | |
| Hematology | ||||||
| Anemia | 72 | < 1 | < 1 | 58 | 0 | 0 |
| Thrombocytopenia | 27 | 5 | < 1 | 24 | 3 | < 1 |
| Neutropenia | 3 | 0 | < 1 | 10 | < 1 | 0 |
| Chemistry | ||||||
| Hypercholesterolemia | 35 | 0 | 0 | 8 | 0 | 0 |
| Elevated ALT | 25 | < 1 | 0 | 16 | 0 | 0 |
| Elevated AST | 23 | 0 | 0 | 23 | < 1 | 0 |
| Hypertriglyceridemia | 15 | 0 | 0 | 13 | 0 | 0 |
| a Presented values are worst Grade values regardless of baseline b National Cancer Institute Common Terminology Criteria for Adverse Events, version 3.0 |
||||||
Acute Graft-Versus-Host Disease
In a single-arm, open-label study, 71 adults (ages 18-73 years) were treated with Jakafi for aGVHD failing treatment with steroids with or without other immunosuppressive drugs [see Clinical Studies]. The median duration of treatment with Jakafi was 46 days (range, 4-382 days).
There were no fatal adverse reactions to Jakafi. An adverse reaction resulting in treatment discontinuation occurred in 31% of patients. The most common adverse reaction leading to treatment discontinuation was infection (10%). Table 16 shows the adverse reactions other than laboratory abnormalities.
Table 16: Acute Graft-Versus-Host Disease: Nonhematologic Adverse Reactions Occurring in ≥ 15% of Patients in the Open-Label, Single-Cohort Study
| Adverse Reactionsa | Jakafi (N=71) |
|
| All Gradesb (%) | Grade 3-4 (%) | |
| Infections (pathogen not specified) | 55 | 41 |
| Edema | 51 | 13 |
| Hemorrhage | 49 | 20 |
| Fatigue | 37 | 14 |
| Bacterial infections | 32 | 28 |
| Dyspnea | 32 | 7 |
| Viral infections | 31 | 14 |
| Thrombosis | 25 | 11 |
| Diarrhea | 24 | 7 |
| Rash | 23 | 3 |
| Headache | 21 | 4 |
| Hypertension | 20 | 13 |
| Dizziness | 16 | 0 |
| a Selected laboratory abnormalities are listed in Table 17 below b National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE), version 4.03 |
||
Selected laboratory abnormalities during treatment with Jakafi are shown in Table 17.
Table 17: Acute Graft-Versus-Host Disease: Selected Laboratory Abnormalities Worsening from Baseline in the Open-Label, Single Cohort Study
| Laboratory Parameter | Jakafi (N=71) |
|
| Worst grade during treatment | ||
| All Gradesa (%) | Grade 3-4 (%) | |
| Hematology | ||
| Anemia | 75 | 45 |
| Thrombocytopenia | 75 | 61 |
| Neutropenia | 58 | 40 |
| Chemistry | ||
| Elevated ALT | 48 | 8 |
| Elevated AST | 48 | 6 |
| Hypertriglyceridemia | 11 | 1 |
| a National Cancer Institute Common Terminology Criteria for Adverse Events, version 4.03 | ||
Chronic Graft-Versus-Host Disease
In a Phase 3, randomized, open-label, multi-center study, 165 patients were treated with Jakafi and 158 patients were treated with best available therapy for cGVHD failing treatment with steroids with or without other immunosuppressive drugs [see Clinical Studies]; sixty-five patients crossed over from best available therapy to treatment with Jakafi, for a total of 230 patients treated with Jakafi. The median duration of exposure to Jakafi for the study was 49.7 weeks (range, 0.7 to 144.9 weeks) in the Jakafi arm. One hundred and nine (47%) patients were on Jakafi for at least 1 year.
There were five fatal adverse reactions to Jakafi, including 1 from toxic epidermal necrolysis and 4 from neutropenia, anemia and/or thrombocytopenia. An adverse reaction resulting in treatment discontinuation occurred in 18% of patients treated with Jakafi. An adverse reaction resulting in dose modification occurred in 27%, and an adverse reaction resulting in treatment interruption occurred in 23%. The most common hematologic adverse reactions (incidence > 35%) are anemia and thrombocytopenia. The most common nonhematologic adverse reactions (incidence ≥ 20%) are infections (pathogen not specified) and viral infection.
Table 18 presents the most frequent nonlaboratory adverse reactions occurring up to Cycle 7 Day 1 of randomized treatment.
Table 18: Chronic Graft-Versus-Host Disease: All-Grade (≥ 10%) and Grades 3-5 (≥ 3%) Nonlaboratory Adverse Reactions Occurring in Patients in the Open-Label, Active-controlled Study up to Cycle 7 Day 1 of Randomized Treatment
| Adverse Reactionsb | Jakafi (N = 165) |
Best Available Therapy (N = 158) |
||
| All Gradesa (%) | Grade ≥ 3 (%) | All Grades (%) | Grade ≥ 3 (%) | |
| Infections and infestations | ||||
| Infections (pathogen not specified) | 45 | 15 | 44 | 16 |
| Viral infections | 28 | 5 | 23 | 5 |
| Musculoskeletal and connective tissue disorders | ||||
| Musculoskeletal pain | 18 | 1 | 13 | 0 |
| General disorders and administration site conditions | ||||
| Pyrexia | 16 | 2 | 9 | 1 |
| Fatigue | 13 | 1 | 10 | 2 |
| Edema | 10 | 1 | 12 | 1 |
| Vascular disorders | ||||
| Hypertension | 16 | 5 | 13 | 7 |
| Hemorrhage | 12 | 2 | 15 | 2 |
| Respiratory, thoracic and mediastinal disorders | ||||
| Cough | 13 | 0 | 8 | 0 |
| Dyspnea | 11 | 1 | 8 | 1 |
| Gastrointestinal disorders | ||||
| Nausea | 12 | 0 | 13 | 2 |
| Diarrhea | 10 | 1 | 13 | 1 |
| a National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE), version 4.03 b Grouped terms that are composites of applicable adverse reaction terms. |
||||
Clinically relevant laboratory abnormalities are shown in Table 19.
Table 19: Chronic Graft-Versus-Host Disease: Selected Laboratory Abnormalities in the Open-Label, Active-controlled Study up to Cycle 7 Day 1 of Randomized Treatmenta
| Laboratory Test | Jakafi (N = 165) |
Best Available Therapy (N = 158) |
||
| All Gradesb (%) | Grade ≥ 3 (%) | All Grades (%) | Grade ≥ 3 (%) | |
| Hematology | ||||
| Anemia | 82 | 13 | 75 | 8 |
| Neutropenia | 27 | 12 | 23 | 9 |
| Thrombocytopenia | 58 | 20 | 54 | 17 |
| Chemistry | ||||
| Hypercholesterolemia | 88 | 10 | 85 | 8 |
| Elevated AST | 65 | 5 | 54 | 6 |
| Elevated ALT | 73 | 11 | 71 | 16 |
| Gamma glutamyltransferase increased | 81 | 42 | 75 | 38 |
| Creatinine increased | 47 | 1 | 40 | 2 |
| Elevated lipase | 38 | 12 | 30 | 9 |
| Elevated amylase | 35 | 8 | 25 | 4 |
| a Presented values are worst Grade values regardless of baseline b National Cancer Institute Common Terminology Criteria for Adverse Events, version 4.03 |
||||
Postmarketing Experience
The following adverse reactions have been identified during post-approval use of Jakafi. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure:
- Infections and Infestations: Herpes simplex virus reactivation and/or dissemination
Drug Interactions for Jakafi
Effect Of Other Drugs On Jakafi
Fluconazole
Concomitant use of Jakafi with fluconazole increases ruxolitinib exposure [see CLINICAL PHARMACOLOGY], which may increase the risk of exposure-related adverse reactions. Avoid concomitant use of Jakafi with fluconazole doses of greater than 200 mg daily. Reduce the Jakafi dosage when used concomitantly with fluconazole doses of less than or equal to 200 mg [see DOSAGE AND ADMINISTRATION].
Strong CYP3A4 Inhibitors
Concomitant use of Jakafi with strong CYP3A4 inhibitors increases ruxolitinib exposure [see CLINICAL PHARMACOLOGY], which may increase the risk of exposure-related adverse reactions. Reduce the Jakafi dosage when used concomitantly with strong CYP3A4 inhibitors except in patients with aGVHD or cGVHD [see DOSAGE AND ADMINISTRATION].
Strong CYP3A4 Inducers
Concomitant use of Jakafi with strong CYP3A4 inducers may decrease ruxolitinib exposure [see CLINICAL PHARMACOLOGY], which may reduce efficacy of Jakafi. Monitor patients frequently and adjust the Jakafi dose based on safety and efficacy [see CLINICAL PHARMACOLOGY].
Warnings for Jakafi
Included as part of the PRECAUTIONS section.
Precautions for Jakafi
Thrombocytopenia, Anemia And Neutropenia
Treatment with Jakafi can cause thrombocytopenia, anemia and neutropenia [see ADVERSE REACTIONS].
Manage thrombocytopenia by reducing the dose or temporarily interrupting Jakafi. Platelet transfusions may be necessary [see DOSAGE AND ADMINISTRATION].
Patients developing anemia may require blood transfusions and/or dose modifications of Jakafi.
Severe neutropenia (ANC less than 0.5 × 109/L) was generally reversible by withholding Jakafi until recovery.
Perform a pre-treatment complete blood count (CBC) and monitor CBCs every 2 to 4 weeks until doses are stabilized, and then as clinically indicated [see DOSAGE AND ADMINISTRATION].
Risk Of Infection
Serious bacterial, mycobacterial, fungal and viral infections have occurred [see ADVERSE REACTIONS]. Delay starting therapy with Jakafi until active serious infections have resolved. Observe patients receiving Jakafi for signs and symptoms of infection and manage promptly. Use active surveillance and prophylactic antibiotics according to clinical guidelines.
Tuberculosis
Tuberculosis infection has been reported in patients receiving Jakafi. Observe patients receiving Jakafi for signs and symptoms of active tuberculosis and manage promptly.
Prior to initiating Jakafi, patients should be evaluated for tuberculosis risk factors, and those at higher risk should be tested for latent infection. Risk factors include, but are not limited to, prior residence in or travel to countries with a high prevalence of tuberculosis, close contact with a person with active tuberculosis, and a history of active or latent tuberculosis where an adequate course of treatment cannot be confirmed.
For patients with evidence of active or latent tuberculosis, consult a physician with expertise in the treatment of tuberculosis before starting Jakafi. The decision to continue Jakafi during treatment of active tuberculosis should be based on the overall risk-benefit determination.
Progressive Multifocal Leukoencephalopathy
Progressive multifocal leukoencephalopathy (PML) has occurred with Jakafi treatment. If PML is suspected, stop Jakafi and evaluate.
Herpes Zoster And Herpes Simplex
Herpes zoster infection has been reported in patients receiving Jakafi [see ADVERSE REACTIONS]. Advise patients about early signs and symptoms of herpes zoster and to seek treatment as early as possible if suspected.
Herpes simplex virus reactivation and/or dissemination has been reported in patients receiving Jakafi [see ADVERSE REACTIONS]. Monitor patients for the development of herpes simplex infections. If a patient develops evidence of dissemination of herpes simplex, consider interrupting treatment with Jakafi; patients should be promptly treated and monitored according to clinical guidelines.
Hepatitis B
Hepatitis B viral load (HBV-DNA titer) increases, with or without associated elevations in alanine aminotransferase and aspartate aminotransferase, have been reported in patients with chronic HBV infections taking Jakafi. The effect of Jakafi on viral replication in patients with chronic HBV infection is unknown. Patients with chronic HBV infection should be treated and monitored according to clinical guidelines.
Symptom Exacerbation Following Interruption Or Discontinuation Of Treatment With Jakafi
Following discontinuation of Jakafi, symptoms from myeloproliferative neoplasms may return to pretreatment levels over a period of approximately one week. Some patients with MF have experienced one or more of the following adverse events after discontinuing Jakafi: fever, respiratory distress, hypotension, DIC, or multi-organ failure. If one or more of these occur after discontinuation of, or while tapering the dose of Jakafi, evaluate for and treat any intercurrent illness and consider restarting or increasing the dose of Jakafi. Instruct patients not to interrupt or discontinue Jakafi therapy without consulting their physician. When discontinuing or interrupting therapy with Jakafi for reasons other than thrombocytopenia or neutropenia [see DOSAGE AND ADMINISTRATION], consider tapering the dose of Jakafi gradually rather than discontinuing abruptly.
Non-Melanoma Skin Cancer (NMSC)
Non-melanoma skin cancers including basal cell, squamous cell, and Merkel cell carcinoma have occurred in patients treated with Jakafi. Perform periodic skin examinations.
Lipid Elevations
Treatment with Jakafi has been associated with increases in lipid parameters including total cholesterol, low-density lipoprotein (LDL) cholesterol, and triglycerides [see ADVERSE REACTIONS]. The effect of these lipid parameter elevations on cardiovascular morbidity and mortality has not been determined in patients treated with Jakafi. Assess lipid parameters approximately 8-12 weeks following initiation of Jakafi therapy. Monitor and treat according to clinical guidelines for the management of hyperlipidemia.
Major Adverse Cardiovascular Events (MACE)
Another JAK-inhibitor has increased the risk of MACE, including cardiovascular death, myocardial infarction, and stroke (compared to those treated with TNF blockers) in patients with rheumatoid arthritis, a condition for which Jakafi is not indicated.
Consider the benefits and risks for the individual patient prior to initiating or continuing therapy with Jakafi particularly in patients who are current or past smokers and patients with other cardiovascular risk factors. Patients should be informed about the symptoms of serious cardiovascular events and the steps to take if they occur.
Thrombosis
Another JAK-inhibitor has increased the risk of thrombosis, including deep venous thrombosis (DVT), pulmonary embolism (PE), and arterial thrombosis (compared to those treated with TNF blockers) in patients with rheumatoid arthritis, a condition for which Jakafi is not indicated. In patients with MF and PV treated with Jakafi in clinical trials, the rates of thromboembolic events were similar in Jakafi and control treated patients.
Patients with symptoms of thrombosis should be promptly evaluated and treated appropriately.
Secondary Malignancies
Another JAK-inhibitor has increased the risk of lymphoma and other malignancies excluding NMSC (compared to those treated with TNF blockers) in patients with rheumatoid arthritis, a condition for which Jakafi is not indicated. Patients who are current or past smokers are at additional increased risk.
Consider the benefits and risks for the individual patient prior to initiating or continuing therapy with Jakafi, particularly in patients with a known secondary malignancy (other than a successfully treated NMSC), patients who develop a malignancy, and patients who are current or past smokers.
Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (PATIENT INFORMATION).
Thrombocytopenia, Anemia And Neutropenia
Inform patients that Jakafi is associated with thrombocytopenia, anemia and neutropenia, and of the need to monitor complete blood counts before and during treatment. Advise patients to observe for and report bleeding [see WARNINGS AND PRECAUTIONS].
Infections
Inform patients of the signs and symptoms of infection and to report any such signs and symptoms promptly.
Inform patients regarding the early signs and symptoms of herpes zoster and of progressive multifocal leukoencephalopathy, and advise patients to seek advice of a clinician if such symptoms are observed [see WARNINGS AND PRECAUTIONS].
Symptom Exacerbation Following Interruption Or Discontinuation Of Treatment With Jakafi
Inform patients that after discontinuation of treatment, signs and symptoms from myeloproliferative neoplasms are expected to return. Instruct patients not to interrupt or discontinue Jakafi therapy without consulting their physician [see WARNINGS AND PRECAUTIONS].
Non-Melanoma Skin Cancer
Inform patients that Jakafi may increase their risk of certain non-melanoma skin cancers. Advise patients to inform their healthcare provider if they have ever had any type of skin cancer or if they observe any new or changing skin lesions [see WARNINGS AND PRECAUTIONS].
Lipid Elevations
Inform patients that Jakafi may increase blood cholesterol, and of the need to monitor blood cholesterol levels [see WARNINGS AND PRECAUTIONS].
Major Adverse Cardiovascular Events (MACE)
Advise patients that events of major adverse cardiovascular events (MACE) including myocardial infarction, stroke, and cardiovascular death, have been reported in clinical studies with another JAK-inhibitor used to treat rheumatoid arthritis, a condition for which Jakafi is not indicated. Advise patients, especially current or past smokers or patients with other cardiovascular risk factors, to be alert for the development of signs and symptoms of cardiovascular events [see WARNINGS AND PRECAUTIONS].
Thrombosis
Advise patients that events of DVT and PE have been reported in clinical studies with another JAK-inhibitor used to treat rheumatoid arthritis, a condition for which Jakafi is not indicated. Advise patients to tell their healthcare provider if they develop any signs or symptoms of a DVT or PE [see WARNINGS AND PRECAUTIONS].
Secondary Malignancies
Advise patients, especially current or past smokers and patients with a known secondary malignancy (other than a successfully treated NMSC), that lymphoma and other malignancies (excluding NMSC) have been reported in clinical studies with another JAK-inhibitor used to treat rheumatoid arthritis, a condition for which Jakafi is not indicated [see WARNINGS AND PRECAUTIONS].
Drug-Drug Interactions
Advise patients to inform their healthcare providers of all medications they are taking, including over-the-counter medications, herbal products and dietary supplements [see DRUG INTERACTIONS and CLINICAL PHARMACOLOGY].
Dialysis
Inform patients on dialysis that their dose should not be taken before dialysis but only following dialysis [see DOSAGE AND ADMINISTRATION].
Lactation
Inform women not to breastfeed during treatment with Jakafi and for two weeks after the final dose [see Use In Specific Populations].
Compliance
Advise patients to continue taking Jakafi every day for as long as their physician tells them and that this is a long-term treatment. Patients should not change dose or stop taking Jakafi without first consulting their physician. Patients should be aware that after discontinuation of treatment, signs and symptoms from myeloproliferative neoplasms are expected to return.
Nonclinical Toxicology
Carcinogenesis, Mutagenesis, Impairment Of Fertility
Ruxolitinib was not carcinogenic in the 6-month Tg.rasH2 transgenic mouse model or in a 2-year carcinogenicity study in the rat.
Ruxolitinib was not mutagenic in a bacterial mutagenicity assay (Ames test) or clastogenic in in vitro chromosomal aberration assay (cultured human peripheral blood lymphocytes) or in vivo in a rat bone marrow micronucleus assay.
In a fertility study, ruxolitinib was administered to male rats prior to and throughout mating and to female rats prior to mating and up to the implantation day (gestation day 7). Ruxolitinib had no effect on fertility or reproductive function in male or female rats at doses of 10, 30 or 60 mg/kg/day. However, in female rats doses of greater than or equal to 30 mg/kg/day resulted in increased post-implantation loss. The exposure (AUC) at the dose of 30 mg/kg/day is approximately 34% the clinical exposure at the maximum recommended dose of 25 mg twice daily.
Use In Specific Populations
Pregnancy
Risk Summary
When pregnant rats and rabbits were administered ruxolitinib during the period of organogenesis adverse developmental outcomes occurred at doses associated with maternal toxicity (see Data). There are no studies with the use of Jakafi in pregnant women to inform drug-associated risks.
The background risk of major birth defects and miscarriage for the indicated populations is unknown. Adverse outcomes in pregnancy occur regardless of the health of the mother or the use of medications. The background risk in the U.S. general population of major birth defects is 2% to 4% and miscarriage is 15% to 20% of clinically recognized pregnancies.
Data
Animal Data
Ruxolitinib was administered orally to pregnant rats or rabbits during the period of organogenesis, at doses of 15, 30 or 60 mg/kg/day in rats and 10, 30 or 60 mg/kg/day in rabbits. There were no treatment-related malformations. Adverse developmental outcomes, such as decreases of approximately 9% in fetal weights were noted in rats at the highest and maternally toxic dose of 60 mg/kg/day. This dose results in an exposure (AUC) that is approximately 2 times the clinical exposure at the maximum recommended dose of 25 mg twice daily. In rabbits, lower fetal weights of approximately 8% and increased late resorptions were noted at the highest and maternally toxic dose of 60 mg/kg/day. This dose is approximately 7% the clinical exposure at the maximum recommended dose.
In a pre-and post-natal development study in rats, pregnant animals were dosed with ruxolitinib from implantation through lactation at doses up to 30 mg/kg/day. There were no drug-related adverse findings in pups for fertility indices or for maternal or embryofetal survival, growth and development parameters at the highest dose evaluated (34% the clinical exposure at the maximum recommended dose of 25 mg twice daily).
Lactation
Risk Summary
No data are available regarding the presence of ruxolitinib in human milk, the effects on the breast fed child, or the effects on milk production. Ruxolitinib and/or its metabolites were present in the milk of lactating rats (see Data). Because many drugs are present in human milk and because of the potential for thrombocytopenia and anemia shown for Jakafi in human studies, discontinue breastfeeding during treatment with Jakafi and for two weeks after the final dose.
Data
Animal Data
Lactating rats were administered a single dose of [14C]-labeled ruxolitinib (30 mg/kg) on postnatal Day 10, after which plasma and milk samples were collected for up to 24 hours. The AUC for total radioactivity in milk was approximately 13-fold the maternal plasma AUC. Additional analysis showed the presence of ruxolitinib and several of its metabolites in milk, all at levels higher than those in maternal plasma.
Pediatric Use
Myelofibrosis
The safety and effectiveness of Jakafi for treatment of myelofibrosis in pediatric patients have not been established.
Polycythemia Vera
The safety and effectiveness of Jakafi for treatment of polycythemia vera in pediatric patients have not been established.
Acute Graft-Versus-Host Disease
The safety and effectiveness of Jakafi for treatment of steroid-refractory aGVHD has been established for treatment of pediatric patients 12 years and older. Use of Jakafi in pediatric patients with steroid-refractory aGVHD is supported by evidence from adequate and well-controlled trials of Jakafi in adults [see Clinical Studies] and additional pharmacokinetic and safety data in pediatric patients. The safety and effectiveness of Jakafi for treatment of steroid-refractory aGVHD has not been established in pediatric patients younger than 12 years old.
Chronic Graft-Versus-Host Disease
The safety and effectiveness of Jakafi for treatment of cGVHD after failure of one or two lines of systemic therapy has been established for treatment of pediatric patients 12 years and older. Use of Jakafi in pediatric patients with cGVHD after failure of one or two lines of systemic therapy is supported by evidence from adequate and well-controlled trials of Jakafi in adults and adolescents [see Clinical Studies] and additional pharmacokinetic and safety data in pediatric patients. The safety and effectiveness of Jakafi for treatment of cGVHD has not been established in pediatric patients younger than 12 years old.
Other Myeloproliferative Neoplasms, Leukemias, And Solid Tumors
The safety and effectiveness of ruxolitinib were assessed but not established in a single-arm trial (NCT01164163) in patients with relapsed or refractory solid tumors, leukemias, or myeloproliferative neoplasms. The patients included 18 children (age 2 to < 12 years) and 14 adolescents (age 12 to < 17 years). Overall, 19% of patients received more than one cycle. No new safety signals were observed in pediatric patients in this trial.
The safety and effectiveness of ruxolitinib in combination with chemotherapy for treatment of high-risk, de novo CRLF2 rearranged or JAK pathway–mutant Ph-like acute lymphoblastic leukemia (ALL) were assessed but not established in a single-arm trial (NCT02723994). The patients included 2 infants (age < 2 years), 42 children (age 2 to < 12 years) and 62 adolescents (age 12 to < 17 years). No new safety signals were observed in pediatric patients in this trial.
Juvenile Animal Toxicity Data
Administration of ruxolitinib to juvenile rats resulted in effects on growth and bone measures. When administered starting at postnatal day 7 (the equivalent of a human newborn) at doses of 1.5 to 75 mg/kg/day, evidence of fractures occurred at doses ≥ 30 mg/kg/day, and effects on body weight and other bone measures [e.g., bone mineral content, peripheral quantitative computed tomography, and x-ray analysis] occurred at doses ≥ 5 mg/kg/day. When administered starting at postnatal day 21 (the equivalent of a human 2-3 years of age) at doses of 5 to 60 mg/kg/day, effects on body weight and bone occurred at doses ≥ 15 mg/kg/day, which were considered adverse at 60 mg/kg/day. Males were more severely affected than females in all age groups, and effects were generally more severe when administration was initiated earlier in the postnatal period. These findings were observed at exposures that are at least 27% the clinical exposure at the maximum recommended dose of 25 mg twice daily.
Geriatric Use
Of the total number of patients with MF in clinical studies with Jakafi, 52% were 65 years and older, while 15% were 75 years and older. No overall differences in safety or effectiveness of Jakafi were observed between these patients and younger patients.
Clinical studies of Jakafi in patients with aGVHD did not include sufficient numbers of subjects age 65 and over to determine whether they respond differently from younger subjects.
Of the total number of patients with cGVHD treated with Jakafi in clinical trials, 11% were 65 years and older. No overall differences in safety or effectiveness of Jakafi were observed between these patients and younger patients.
Renal Impairment
Total exposure of ruxolitinib and its active metabolites increased with moderate (CLcr 30 to 59 mL/min) and severe (CLcr 15 to 29 mL/min) renal impairment, and ESRD (CLcr less than 15 mL/min) on dialysis [see CLINICAL PHARMACOLOGY]. Modify Jakafi dosage as recommended [see DOSAGE AND ADMINISTRATION].
Hepatic Impairment
Exposure of ruxolitinib increased with mild (Child-Pugh A), moderate (Child-Pugh B) and severe (Child-Pugh C) hepatic impairment [see CLINICAL PHARMACOLOGY].
Reduce Jakafi dosage as recommended in patients with MF or PV with hepatic impairment [see DOSAGE AND ADMINISTRATION]. Reduce Jakafi dosage as recommended for patients with Stage 4 liver aGVHD.
Monitor blood counts more frequently for toxicity and modify the Jakafi dosage for adverse reactions if they occur for patients with Score 3 liver cGVHD [see DOSAGE AND ADMINISTRATION and CLINICAL PHARMACOLOGY].
Overdose Information for Jakafi
There is no known antidote for overdoses with Jakafi. Single doses up to 200 mg have been given with acceptable acute tolerability. Higher than recommended repeat doses are associated with increased myelosuppression including leukopenia, anemia and thrombocytopenia. Appropriate supportive treatment should be given.
Hemodialysis is not expected to enhance the elimination of Jakafi.
Contraindications for Jakafi
None.
Clinical Pharmacology for Jakafi
Mechanism Of Action
Ruxolitinib, a kinase inhibitor, inhibits Janus Associated Kinases (JAKs) JAK1 and JAK2 which mediate the signaling of a number of cytokines and growth factors that are important for hematopoiesis and immune function. JAK signaling involves recruitment of STATs (signal transducers and activators of transcription) to cytokine receptors, activation and subsequent localization of STATs to the nucleus leading to modulation of gene expression.
MF and PV are myeloproliferative neoplasms (MPN) known to be associated with dysregulated JAK1 and JAK2 signaling. In a mouse model of JAK2V617F-positive MPN, oral administration of ruxolitinib prevented splenomegaly, preferentially decreased JAK2V617F mutant cells in the spleen and decreased circulating inflammatory cytokines (e.g., TNF-α, IL-6).
JAK-STAT signaling pathways play a role in regulating the development, proliferation, and activation of several immune cell types important for GVHD pathogenesis. In a mouse model of aGVHD, oral administration of ruxolitinib was associated with decreased expression of inflammatory cytokines in colon homogenates and reduced immune-cell infiltration in the colon.
Pharmacodynamics
Jakafi inhibits cytokine induced STAT3 phosphorylation in whole blood from patients with MF and PV. STAT3 phosphorylation reached maximal inhibition 2 hours after Jakafi dosing and returned to near baseline by 10 hours in patients with MF and PV.
Cardiac Electrophysiology
At a dose of 1.25 to 10 times the highest recommended starting dosage, Jakafi does not prolong the QT interval to any clinically relevant extent.
Pharmacokinetics
Mean ruxolitinib maximal plasma concentration (Cmax) and AUC increased proportionally over a single dose range of 5 mg to 200 mg (4 times the approved highest recommended total daily dosage of 25 mg twice daily). Mean ruxolitinib Cmax ranged from 205 nM to 7100 nM and AUC ranged from 862 nM*hr to 30700 nM*hr over a single dose range of 5 mg to 200 mg.
Absorption
Ruxolitinib achieves Cmax within 1 hour to 2 hours post-dose. Oral absorption of ruxolitinib is estimated to be at least 95%.
Effect Of Food
No clinically relevant changes in the pharmacokinetics of ruxolitinib were observed upon administration of Jakafi with a high-fat, high-calorie meal (approximately 800 to 1000 calories of which 50% were derived from fat).
Distribution
The mean ruxolitinib volume of distribution at steady-state is 72 L (coefficient of variation [CV] 29%) in patients with MF and 75 L (23%) in patients with PV.
Protein binding of ruxolitinib is approximately 97%, mostly to albumin.
Elimination
The mean elimination half-life of ruxolitinib is approximately 3 hours and the mean elimination half-life of ruxolitinib and its metabolites is approximately 5.8 hours in healthy volunteers.
Ruxolitinib clearance (%CV) was 17.7 L/h in women and 22.1 L/h in men with MF (39%).
Ruxolitinib clearance (%CV) was 12.7 L/h (42%) in patients with PV.
Ruxolitinib clearance (%CV) was 11.8 L/h (63%) in patients with aGVHD.
Ruxolitinib clearance (%CV) was 9.7 L/h (51%) in patients with cGVHD.
Metabolism
Ruxolitinib is metabolized by CYP3A4 and to a lesser extent by CYP2C9.
Excretion
Following a single oral dose of radiolabeled ruxolitinib, 74% of radioactivity was excreted in urine and 22% via feces. Unchanged drug accounted for less than 1% of the excreted total radioactivity.
Specific Populations
No clinically relevant differences in ruxolitinib pharmacokinetics were observed based on age (12-73 years), race (White, Asian), sex, or weight (29-139 kg).
Patients With Renal Impairment
Total AUC of ruxolitinib and its active metabolites increased by 1.3-, 1.5-, 1.9-, and 1.6-fold in subjects with mild, moderate, severe renal impairment, and with ESRD after dialysis, respectively, compared to subjects with normal renal function (CLcr ≥ 90 mL/min). The change in the pharmacodynamic marker, pSTAT3 inhibition, was consistent with the corresponding increase in metabolite exposure with renal impairment. Ruxolitinib is not removed by dialysis; however, the removal of some active metabolites by dialysis cannot be ruled out.
Patients With Hepatic Impairment
No clinically relevant effect on ruxolitinib pharmacokinetics was observed based on mild to severe hepatic impairment by NCI criteria (total bilirubin > ULN and any AST) in patients with aGVHD or cGVHD.
Ruxolitinib AUC increased in subjects with mild (Child-Pugh A) by 1.9-fold, moderate (Child-Pugh B) by 1.3-fold, and severe (Child-Pugh C) hepatic impairment by 1.7-fold compared to that in subjects with normal hepatic function.
The change in the pharmacodynamic marker, pSTAT3 inhibition, was consistent with the corresponding increase in ruxolitinib exposure except in the severe hepatic impairment cohort where the pharmacodynamic activity was more prolonged in some subjects than expected based on plasma concentrations of ruxolitinib.
Patients With Liver Involvement In Graft-Versus-Host Disease
No clinically relevant effect on ruxolitinib pharmacokinetics was observed based on Stage 1, 2 or 3 liver aGVHD, or Score 1, or 2 liver cGVHD.
A lower apparent clearance of ruxolitinib was observed in patients with Stage 4 liver aGVHD compared to patients with no liver aGVHD.
The effect of Score 3 liver cGVHD on the pharmacokinetics of ruxolitinib is unknown.
Drug Interaction Studies
Clinical Studies And Model-Informed Approaches
Fluconazole
Fluconazole 100 to 400 mg once daily (a moderate CYP3A4 and CYP2C9 inhibitor) increases steady state ruxolitinib AUC by approximately 100% to 300% [see DOSAGE AND ADMINISTRATION and DRUG INTERACTIONS].
Strong CYP3A4 Inhibitors
Ketoconazole (strong CYP3A4 inhibitor) increased ruxolitinib Cmax by 33% and AUC by 91% and prolonged ruxolitinib half-life from 3.7 hours to 6 hours [see DOSAGE AND ADMINISTRATION and DRUG INTERACTIONS].
Moderate CYP3A4 Inhibitors
Erythromycin (moderate CYP3A4 inhibitor) increased ruxolitinib Cmax by 8% and AUC by 27% [see DRUG INTERACTIONS].
Strong CYP3A4 Inducers
Rifampin (strong CYP3A4 inducer) decreased ruxolitinib Cmax by 32% and AUC by 61%. The relative exposure to ruxolitinib’s active metabolites increased approximately 100% [see DRUG INTERACTIONS].
In Vitro Studies
Cytochrome P450 (CYP) Enzymes
Ruxolitinib and its M18 metabolite did not inhibit CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 or CYP3A4. Ruxolitinib did not induce CYP1A2, CYP2B6 or CYP3A4 at clinically relevant concentrations.
Transporter Systems
Ruxolitinib and its M18 metabolite did not inhibit the P-gp, BCRP, OATP1B1, OATP1B3, OCT1, OCT2, OAT1 or OAT3 at clinically relevant concentrations. Ruxolitinib was not a P-gp substrate.
Clinical Studies
Myelofibrosis
Two randomized Phase 3 studies (Studies 1 and 2) were conducted in patients with MF (either primary MF, post-polycythemia vera MF or post-essential thrombocythemia-MF). In both studies, patients had palpable splenomegaly at least 5 cm below the costal margin and risk category of intermediate 2 (2 prognostic factors) or high risk (3 or more prognostic factors) based on the International Working Group Consensus Criteria (IWG).
The starting dose of Jakafi was based on platelet count. Patients with a platelet count between 100 and 200 × 109/L were started on Jakafi 15 mg twice daily and patients with a platelet count greater than 200 × 109/L were started on Jakafi 20 mg twice daily. Doses were then individualized based upon tolerability and efficacy with maximum doses of 20 mg twice daily for patients with platelet counts between 100 to less than or equal to 125 × 109/L, of 10 mg twice daily for patients with platelet counts between 75 to less than or equal to 100 × 109/L, and of 5 mg twice daily for patients with platelet counts between 50 to less than or equal to 75 × 109/L.
Study 1
Study 1 (NCT00952289) was a double-blind, randomized, placebo-controlled study in 309 patients who were refractory to or were not candidates for available therapy. The median age was 68 years (range 40 to 91 years) with 61% of patients older than 65 years and 54% were male. Fifty percent (50%) of patients had primary MF, 31% had post-polycythemia vera MF and 18% had post-essential thrombocythemia MF. Twenty-one percent (21%) of patients had red blood cell transfusions within 8 weeks of enrollment in the study. The median hemoglobin count was 10.5 g/dL and the median platelet count was 251 × 109/L. Patients had a median palpable spleen length of 16 cm below the costal margin, with 81% having a spleen length 10 cm or greater below the costal margin. Patients had a median spleen volume as measured by magnetic resonance imaging (MRI) or computed tomography (CT) of 2595 cm3 (range 478 cm3 to 8881 cm3). (The upper limit of normal is approximately 300 cm3).
Patients were dosed with Jakafi or matching placebo. The primary efficacy endpoint was the proportion of patients achieving greater than or equal to a 35% reduction from baseline in spleen volume at Week 24 as measured by MRI or CT.
Secondary endpoints included duration of a 35% or greater reduction in spleen volume and proportion of patients with a 50% or greater reduction in Total Symptom Score from baseline to Week 24 as measured by the modified Myelofibrosis Symptom Assessment Form (MFSAF) v2.0 diary.
Study 2
Study 2 (NCT00934544) was an open-label, randomized study in 219 patients. Patients were randomized 2:1 to Jakafi versus best available therapy. Best available therapy was selected by the investigator on a patient-by-patient basis. In the best available therapy arm, the medications received by more than 10% of patients were hydroxyurea (47%) and glucocorticoids (16%). The median age was 66 years (range 35 to 85 years) with 52% of patients older than 65 years and 57% were male. Fifty-three percent (53%) of patients had primary MF, 31% had postÂpolycythemia vera MF and 16% had post-essential thrombocythemia MF. Twenty-one percent (21%) of patients had red blood cell transfusions within 8 weeks of enrollment in the study. The median hemoglobin count was 10.4 g/dL and the median platelet count was 236 × 109/L. Patients had a median palpable spleen length of 15 cm below the costal margin, with 70% having a spleen length 10 cm or greater below the costal margin. Patients had a median spleen volume as measured by MRI or CT of 2381 cm3 (range 451 cm3 to 7765 cm3).
The primary efficacy endpoint was the proportion of patients achieving 35% or greater reduction from baseline in spleen volume at Week 48 as measured by MRI or CT.
A secondary endpoint in Study 2 was the proportion of patients achieving a 35% or greater reduction of spleen volume as measured by MRI or CT from baseline to Week 24.
Study 1 And 2 Efficacy Results
Efficacy analyses of the primary endpoint in Studies 1 and 2 are presented in Table 20 below. A significantly larger proportion of patients in the Jakafi group achieved a 35% or greater reduction in spleen volume from baseline in both studies compared to placebo in Study 1 and best available therapy in Study 2. A similar proportion of patients in the Jakafi group achieved a 50% or greater reduction in palpable spleen length.
Table 20: Percent of Patients with Myelofibrosis Achieving 35% or Greater Reduction from Baseline in Spleen Volume at Week 24 in Study 1 and at Week 48 in Study 2 (Intent to Treat)
| Study 1 | Study 2 | |||
| Jakafi (N=155) |
Placebo (N=154) |
Jakafi (N=146) |
Best Available Therapy (N=73) |
|
| Time Points | Week 24 | Week 48 | ||
| Number (%) of Patients with Spleen Volume Reduction by 35% or More | 65 (42) | 1 (< 1) | 41 (29) | 0 |
| P-value | < 0.0001 | < 0.0001 | ||
Figure 1 shows the percent change from baseline in spleen volume for each patient at Week 24 (Jakafi N=139, placebo N=106) or the last evaluation prior to Week 24 for patients who did not complete 24 weeks of randomized treatment (Jakafi N=16, placebo N=47). One (1) patient (placebo) with a missing baseline spleen volume is not included.
Figure 1: Percent Change from Baseline in Spleen Volume at Week 24 or Last Observation for Each Patient (Study 1)
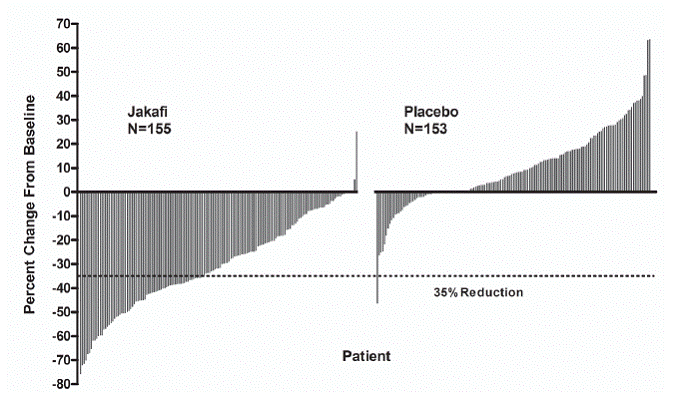 |
In Study 1, MF symptoms were a secondary endpoint and were measured using the modified Myelofibrosis Symptom Assessment Form (MFSAF) v2.0 diary. The modified MFSAF is a daily diary capturing the core symptoms of MF (abdominal discomfort, pain under left ribs, night sweats, itching, bone/muscle pain and early satiety). Symptom scores ranged from 0 to 10 with 0 representing symptoms “absent” and 10 representing “worst imaginable” symptoms. These scores were added to create the daily total score, which has a maximum of 60.
Table 21 presents assessments of Total Symptom Score from baseline to Week 24 in Study 1 including the proportion of patients with at least a 50% reduction (ie, improvement in symptoms). At baseline, the mean Total Symptom Score was 18.0 in the Jakafi group and 16.5 in the placebo group. A higher proportion of patients in the Jakafi group had a 50% or greater reduction in Total Symptom Score than in the placebo group, with a median time to response of less than 4 weeks.
Table 21: Improvement in Total Symptom Score in Patients with Myelofibrosis
| Jakafi (N=148) |
Placebo (N=152) |
|
| Number (%) of Patients with 50% or Greater Reduction in Total Symptom Score by Week 24 | 68 (46) | 8 (5) |
| P-value | < 0.0001 | |
Figure 2 shows the percent change from baseline in Total Symptom Score for each patient at Week 24 (Jakafi N=129, placebo N=103) or the last evaluation on randomized therapy prior to Week 24 for patients who did not complete 24 weeks of randomized treatment (Jakafi N=16, placebo N=42). Results are excluded for 5 patients with a baseline Total Symptom Score of zero, 8 patients with missing baseline and 6 patients with insufficient post-baseline data.
Figure 2: Percent Change from Baseline in Total Symptom Score at Week 24 or Last Observation for Each Patient (Study 1)
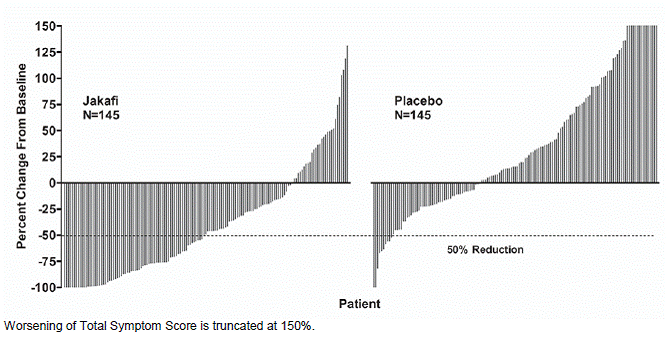 |
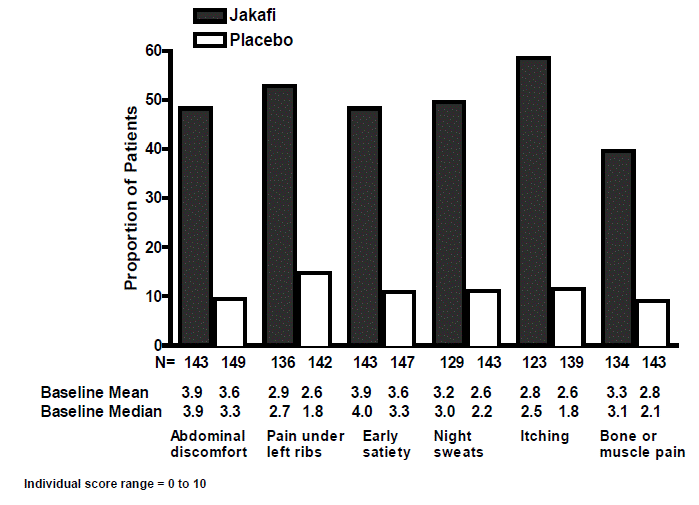
Figure 3 displays the proportion of patients with at least a 50% improvement in each of the individual symptoms that comprise the Total Symptom Score indicating that all 6 of the symptoms contributed to the higher Total Symptom Score response rate in the group treated with Jakafi.
Figure 3: Proportion of Patients with Myelofibrosis Achieving 50% or Greater Reduction in Individual Symptom Scores at Week 24
 |
An exploratory analysis of patients receiving Jakafi also showed improvement in fatigue-related symptoms (i.e., tiredness, exhaustion, mental tiredness, and lack of energy) and associated impacts on daily activities (i.e., activity limitations related to work, self-care, and exercise) as measured by the PROMIS® Fatigue 7-item short form total score at Week 24. Patients who achieved a reduction of 4.5 points or more from baseline to Week 24 in the PROMIS® Fatigue total score were considered to have achieved a fatigue response. Fatigue response was reported in 35% of patients in the Jakafi group versus 14% of the patients in the placebo group.
Overall survival was a secondary endpoint in both Study 1 and Study 2. Patients in the control groups were eligible for crossover in both studies, and the median times to crossover were 9 months in Study 1 and 17 months in Study 2.
Figure 4 and Figure 5 show Kaplan-Meier curves of overall survival at prospectively planned analyses after all patients remaining on study had completed 144 weeks on study.
Figure 4: Overall Survival -Kaplan-Meier Curves by Treatment Group in Study 1
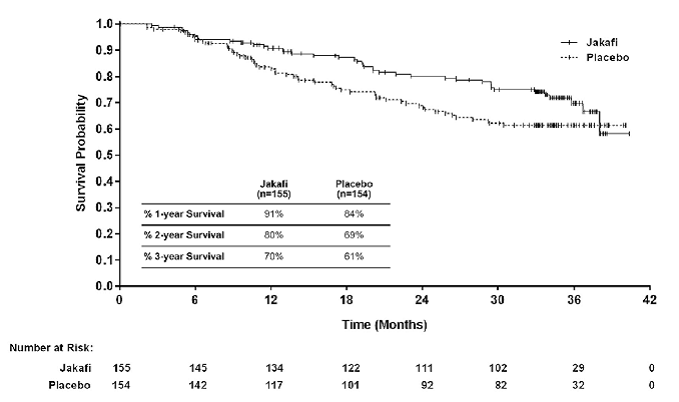 |
Figure 5: Overall Survival -Kaplan-Meier Curves by Treatment Group in Study 2
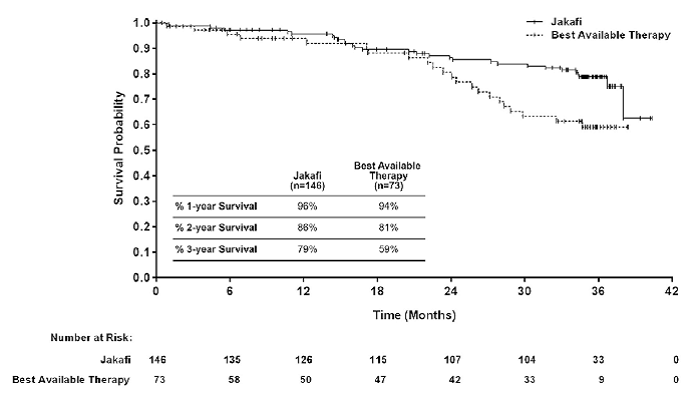 |
Polycythemia Vera
Study 3 (NCT01243944) was a randomized, open-label, active-controlled Phase 3 study conducted in 222 patients with PV. Patients had been diagnosed with PV for at least 24 weeks, had an inadequate response to or were intolerant of hydroxyurea, required phlebotomy and exhibited splenomegaly. All patients were required to demonstrate hematocrit control between 40-45% prior to randomization. The age ranged from 33 to 90 years with 30% of patients over 65 years of age and 66% were male. Patients had a median spleen volume as measured by MRI or CT of 1272 cm3 (range 254 cm3 to 5147 cm3) and median palpable spleen length below the costal margin was 7 cm.
Patients were randomized to Jakafi or best available therapy. The starting dose of Jakafi was 10 mg twice daily. Doses were then individualized based upon tolerability and efficacy with a maximum dose of 25 mg twice daily. At Week 32, 98 patients were still on Jakafi with 8% receiving greater than 20 mg twice daily, 15% receiving 20 mg twice daily, 33% receiving 15 mg twice daily, 34% receiving 10 mg twice daily, and 10% receiving less than 10 mg twice daily. Best available therapy (BAT) was selected by the investigator on a patient-by-patient basis and included hydroxyurea (60%), interferon/pegylated interferon (12%), anagrelide (7%), pipobroman (2%), lenalidomide/thalidomide (5%), and observation (15%).
The primary endpoint was the proportion of subjects achieving a response at Week 32, with response defined as having achieved both hematocrit control (the absence of phlebotomy eligibility beginning at the Week 8 visit and continuing through Week 32) and spleen volume reduction (a greater than or equal to 35% reduction from baseline in spleen volume at Week 32). Phlebotomy eligibility was defined as a confirmed hematocrit greater than 45% that is at least 3 percentage points higher than the hematocrit obtained at baseline or a confirmed hematocrit greater than 48%, whichever was lower. Secondary endpoints included the proportion of all randomized subjects who achieved the primary endpoint and who maintained their response 48 weeks after randomization, and the proportion of subjects achieving complete hematological remission at Week 32 with complete hematological remission defined as achieving hematocrit control, platelet count less than or equal to 400 × 109/L, and white blood cell count less than or equal to 10 × 109/L.
Results of the primary and secondary endpoints are presented in Table 22. A significantly larger proportion of patients on the Jakafi arm achieved a response for the primary endpoint compared to best available therapy at Week 32 and maintained their response 48 weeks after randomization. A significantly larger proportion of patients on the Jakafi arm compared to best available therapy also achieved complete hematological remission at Week 32.
Table 22: Percent of Patients with Polycythemia Vera Achieving the Primary and Key Secondary Endpoints in Study 3 (Intent to Treat)
| Jakafi (N=110) |
Best Available Therapy (N=112) |
|
| Number (%) of Patients Achieving a Primary Response at Week 32 | 25 (23%) | 1 (< 1%) |
| 95% CI of the response rate (%) | (15%, 32%) | (0%, 5%) |
| P-value | < 0.0001 | |
| Number (%) of Patients Achieving a Durable Primary Response at Week 48 | 22 (20%) | 1 (< 1%) |
| 95% CI of the response rate (%) | (13%, 29%) | (0%, 5%) |
| P-value | < 0.0001 | |
| Number (%) of Patients Achieving Complete Hematological Remission at Week 32 | 26 (24%) | 9 (8%) |
| 95% CI of the response rate (%) | (16%, 33%) | (4%, 15%) |
| P-value | 0.0016 | |
| Primary Response defined as having achieved both the absence of phlebotomy eligibility beginning at the Week 8 visit and continuing through Week 32 and a greater than or equal to 35% reduction from baseline in spleen volume at Week 32. | ||
Additional analyses for Study 3 to assess durability of response were conducted at Week 80 only in the Jakafi arm. On this arm, 91 (83%) patients were still on treatment at the time of the Week 80 data cut-off. Of the 25 patients who achieved a primary response at Week 32, 19 (76% of the responders) maintained their response through Week 80, and of the 26 patients who achieved complete hematological remission at Week 32, 15 (58% of the responders) maintained their response through Week 80.
In an assessment of the individual components that make up the primary endpoint, there were 66 (60%) patients with hematocrit control on the Jakafi arm vs. 21 (19%) patients on best available therapy at Week 32; 51 (77% of hematocrit responders) patients on the Jakafi arm maintained hematocrit control through Week 80. There were 44 (40%) patients with spleen volume reduction from baseline greater than or equal to 35% on the Jakafi arm vs. 1 (< 1%) patient on best available therapy at Week 32; 43 (98% of spleen volume reduction responders) patients on the Jakafi arm maintained spleen volume reduction through Week 80.
Acute Graft-Versus-Host Disease
Study 4 (NCT02953678) was an open-label, single-arm, multicenter study of Jakafi for treatment of patients with steroid-refractory aGVHD Grades 2 to 4 (Mount Sinai Acute GVHD International Consortium (MAGIC) criteria) occurring after allogeneic hematopoietic stem cell transplantation. Jakafi was administered at 5 mg twice daily, and the dose could be increased to 10 mg twice daily after 3 days in the absence of toxicity.
There were 49 patients with aGVHD refractory to steroids alone. These patients had a median age of 57 years (range, 18-72 years), 47% were male, 92% were Caucasian, and 14% were Hispanic. At baseline, aGVHD was Grade 2 in 27%, Grade 3 in 55%, and Grade 4 in 18%; 84% had visceral GVHD; the median MAGIC biomarker score was 0.47 (range, 0.10-0.92); and the median ST2 level was 334 mcg/L (range, 55-1286 mcg/L). The median duration of prior corticosteroid exposure at baseline was 15 days (range: 3 – 106 days).
The efficacy of Jakafi was based on Day-28 overall response rate (ORR) (complete response, very good partial response or partial response by Center for International Blood and Marrow Transplant Research (CIBMTR) criteria) and the duration of response. The ORR results are presented in Table 23; Day-28 ORR was 100% for Grade 2 GVHD, 40.7% for Grade 3 GVHD, and 44.4% for Grade 4 GVHD.
The median duration of response, calculated from Day-28 response to progression, new salvage therapy for aGVHD or death from any cause (with progression being defined as worsening by one stage in any organ without improvement in other organs in comparison to prior response assessment) was 16 days (95% CI 9, 83). Also, for the Day-28 responders, the median time from Day-28 response to either death or need for new therapy for aGVHD (additional salvage therapy or increase in steroids) was 173 days (95% CI 66, NE).
Table 23: Day-28 Overall Response Rate for Patients with Steroid-Refractory Acute GVHD in Study 4
| Refractory to Steroids Alone (n=49) |
|
| Overall Response (%) (95% CI) | 28 (57.1%) (42.2, 71.2) |
| Complete Response | 15 (30.6%) |
| Very Good Partial Response | 2 (4.1%) |
| Partial Response | 11 (22.4%) |
Chronic Graft-Versus-Host Disease
Study 5 (REACH-3; NCT03112603) was a randomized, open-label, multicenter study of Jakafi in comparison to best available therapy (BAT) for treatment of corticosteroid-refractory cGVHD after allogeneic stem cell transplantation. Eligible patients were ≥ 12 years old with moderate or severe cGVHD as defined by NIH Consensus Criteria requiring additional therapy after failure of corticosteroid therapy and no more than one additional salvage treatment. Patients were excluded if they had ANC < 1 Gi/L and platelet count < 25 Gi/L, estimated creatinine clearance < 30 ml/min, progressive onset cGVHD, oxygen saturation < 90%, total bilirubin > 2 mg/dL, or diarrhea due to GVHD.
A total of 329 patients were randomized 1:1 to receive either Jakafi 10 mg twice daily (n=165) or BAT (n=164). BAT was selected by the investigator prior to randomization and included the following treatments: extracorporeal photopheresis (ECP), low-dose methotrexate (MTX), mycophenolate mofetil (MMF), mTOR inhibitors (everolimus or sirolimus), infliximab, rituximab, pentostatin, imatinib, or ibrutinib. Randomization was stratified by cGVHD severity (moderate versus severe). On Cycle 7 Day 1 and thereafter, patients randomized to BAT could cross over to Jakafi if they had disease progression, mixed response, unchanged response, cGVHD flare, or toxicity to BAT. All patients also received standard supportive care, including anti-infective medications. GVHD prophylaxis and cGVHD treatment medications initiated before randomization, including systemic corticosteroids, calcineurin inhibitors, and topical or inhaled corticosteroid therapy, were allowed to be continued per institutional guidelines. Table 24 shows the demographics and baseline disease characteristics of the randomized population.
Table 24: REACH-3: Demographics and Baseline Chronic GVHD Characteristics
| Jakafi (N=165) |
Best Available Therapy (N=164) |
|
| Median Age, Years (range) | 49 (13, 73) | 50 (12, 76) |
| Age 12 to < 18 Year, n (%) | 4 (2) | 8 (5) |
| Age > 65 Years, n (%) | 18 (11) | 22 (13) |
| Male, n (%) | 109 (66) | 92 (56) |
| Race, n (%) | ||
| White | 116(70) | 132 (81) |
| Black | 2 (1) | 0 |
| Asian | 33 (20) | 21 (13) |
| American Indian or Alaska native | 2 (1) | 0 |
| Other | 9 (6) | 4 (2) |
| Unknown | 3 (2) | 7 (4) |
| Median (range) time (days) from cGVHD diagnosis to randomization | 174 (7-2017) | 150 (10-1947) |
| Prior Therapy | ||
| No prior treatment for cGVHD | 2 (1) | 1 (1) |
| Failed first-line steroids alone | 115 (70) | 125 (76) |
| Failed first-line combination including steroids | 42 (25) | 30 (18) |
| Failed two lines of therapy | 6 (4) | 8 (5) |
| ≥ 4 Organs involved, n (%) | 67 (41) | 63 (38) |
| Severe cGVHD, n (%) | 86 (52) | 79 (48) |
| Median (range) cGVHD Total Symptom Score | 19 (0-80) | 18 (1-54) |
| Median (range) corticosteroid dose at baseline (PE mg/kg)a | 0.29 (0.01-1.81) | 0.26 (0.06-1.21) |
| a Prednisone equivalent milligrams/kilogram | ||
The efficacy of Jakafi was based on overall response rate (ORR) through Cycle 7 Day 1, where overall response included complete response or partial response according to the 2014 NIH Response Criteria and durability of the response. The ORR results are presented in Table 25; the difference in ORR between Jakafi and BAT arms was 13% (95% CI 3%, 23%). The median time to first response in the responders was 3 weeks (range, 2 to 24) for the Jakafi arm and 4 weeks (range, 2 to 25) for the BAT arm. The median duration of response, calculated from first response to progression, death, or new systemic therapies for cGVHD, was 4.2 months (95% CI 3.2, 6.7) for the Jakafi arm and 2.1 months (95% CI 1.6, 3.2) for the BAT arm; and the median time from first response to death or new systemic therapies for cGVHD was 25 months (95% CI 16.8, NE) for the Jakafi arm and 5.6 months (95% CI 4.1, 7.8) for the BAT arm.
Table 25: Overall Response Rate through Cycle 7 Day 1 for Patients with Chronic GVHD in Study 5
| Jakafi (N=165) |
Best Available Therapy (N=164) |
|
| Overall Response (%) | 116 (70%) | 94 (57%) |
| (95% CI)a | (63%, 77%) | (49%, 65%) |
| Complete Response (%) | 14 (8%) | 8 (5%) |
| Partial Response (%) | 102 (62%) | 86 (52%) |
| a 95% CI of Overall Response Rate is estimated using Clopper-Pearson method. | ||
ORR results were supported by exploratory analyses of patient-reported symptom severity which showed at least a 7-point decrease in the cGVHD Total Symptom Score at any time through Cycle 7 Day 1 in 66 (40%; 95% CI 32, 48) patients in the Jakafi arm and 47 (29%; 95% CI 22, 36) patients in the BAT arm.
From 
Report Problems to the Food and Drug Administration
You are encouraged to report negative side effects of prescription drugs to the FDA. Visit the FDA MedWatch website or call 1-800-FDA-1088.