WARNING
LEFT VENTRICULAR DYSFUNCTION and EMBRYO-FETAL TOXICITY
- Left Ventricular Dysfunction: PERJETA can result in subclinical and clinical cardiac failure manifesting as decreased LVEF and CHF. Evaluate cardiac function prior to and during treatment. Discontinue PERJETA treatment for a confirmed clinically significant decrease in left ventricular function [see DOSAGE AND ADMINISTRATION, WARNINGS AND PRECAUTIONS and ADVERSE REACTIONS].
- Embryo-fetal Toxicity: Exposure to PERJETA can result in embryo-fetal death and birth defects. Advise patients of these risks and the need for effective contraception [see WARNINGS AND PRECAUTIONS and Use In Specific Populations].
Description for Perjeta
Pertuzumab is a recombinant humanized monoclonal antibody that targets the extracellular dimerization domain (Subdomain II) of the human epidermal growth factor receptor 2 protein (HER2). Pertuzumab is produced by recombinant DNA technology in a mammalian cell (Chinese Hamster Ovary) culture that may contain the antibiotic, gentamicin. Gentamicin is not detectable in the final product. Pertuzumab has an approximate molecular weight of 148 kDa.
PERJETA injection is a sterile, clear to slightly opalescent, colorless to pale brown liquid for intravenous infusion. Each single use vial contains 420 mg of pertuzumab at a concentration of 30 mg/mL in 20 mM L-histidine acetate (pH 6.0), 120 mM sucrose and 0.02% polysorbate 20.
Uses for Perjeta
Metastatic Breast Cancer (MBC)
PERJETA is indicated for use in combination with trastuzumab and docetaxel for the treatment of patients with HER2-positive metastatic breast cancer who have not received prior anti-HER2 therapy or chemotherapy for metastatic disease [see DOSAGE AND ADMINISTRATION and Clinical Studies].
Early Breast Cancer (EBC)
PERJETA is indicated for use in combination with trastuzumab and chemotherapy for
- the neoadjuvant treatment of patients with HER2-positive, locally advanced, inflammatory, or early stage breast cancer (either greater than 2 cm in diameter or node positive) as part of a complete treatment regimen for early breast cancer [see DOSAGE AND ADMINISTRATION and Clinical Studies].
- the adjuvant treatment of patients with HER2-positive early breast cancer at high risk of recurrence [see DOSAGE AND ADMINISTRATION and Clinical Studies].
Dosage for Perjeta
Patient Selection
Select patients based on HER2 protein overexpression or HER2 gene amplification in tumor specimens [see INDICATIONS AND USAGE and Clinical Studies]. Assessment of HER2 protein overexpression and HER2 gene amplification should be performed using FDA-approved tests specific for breast cancer by laboratories with demonstrated proficiency. Information on the FDA-approved tests for the detection of HER2 protein overexpression and HER2 gene amplification is available at: http://www.fda.gov/CompanionDiagnostics.
Improper assay performance, including use of suboptimally fixed tissue, failure to utilize specified reagents, deviation from specific assay instructions, and failure to include appropriate controls for assay validation, can lead to unreliable results.
Recommended Doses And Schedules
The initial dose of PERJETA is 840 mg administered as a 60-minute intravenous infusion, followed every 3 weeks by a dose of 420 mg administered as an intravenous infusion over 30 to 60 minutes.
When administered with PERJETA, the recommended initial dose of trastuzumab is 8 mg/kg administered as a 90-minute intravenous infusion, followed every 3 weeks by a dose of 6 mg/kg administered as an intravenous infusion over 30 to 90 minutes.
When administered with PERJETA, the recommended initial dose of trastuzumab hyaluronidaseoysk is 600 mg/10,000 units (600 mg trastuzumab and 10,000 units hyaluronidase) administered subcutaneously over approximately 2 to 5 minutes once every three weeks irrespective of the patient’s body weight.
PERJETA, trastuzumab or trastuzumab hyaluronidase-oysk, and taxane should be administered sequentially. PERJETA and trastuzumab or trastuzumab hyaluronidase-oysk can be given in any order. Taxane should be administered after PERJETA and trastuzumab or trastuzumab hyaluronidase-oysk. An observation period of 30 to 60 minutes is recommended after each PERJETA infusion and before commencement of any subsequent administration of trastuzumab or trastuzumab hyaluronidase-oysk, or taxane [see WARNINGS AND PRECAUTIONS].
In patients receiving an anthracycline-based regimen, PERJETA and trastuzumab or trastuzumab hyaluronidase-oysk should be administered following completion of the anthracycline.
Metastatic Breast Cancer (MBC)
When administered with PERJETA, the recommended initial dose of docetaxel is 75 mg/m² administered as an intravenous infusion. The dose may be escalated to 100 mg/m² administered every 3 weeks if the initial dose is well tolerated.
Neoadjuvant Treatment Of Breast Cancer
PERJETA should be administered every 3 weeks for 3 to 6 cycles as part of one of the following treatment regimens for early breast cancer [see Clinical Studies]:
- Four preoperative cycles of PERJETA in combination with trastuzumab or trastuzumab hyaluronidase-oysk and docetaxel followed by 3 postoperative cycles of fluorouracil, epirubicin, and cyclophosphamide (FEC) as given in NeoSphere
- Three or four preoperative cycles of FEC alone followed by 3 or 4 preoperative cycles of PERJETA in combination with docetaxel and trastuzumab or trastuzumab hyaluronidase-oysk as given in TRYPHAENA and BERENICE, respectively
- Six preoperative cycles of PERJETA in combination with docetaxel, carboplatin, and trastuzumab (TCH) or trastuzumab hyaluronidase-oysk (escalation of docetaxel above 75 mg/m² is not recommended) as given in TRYPHAENA
- Four preoperative cycles of dose-dense doxorubicin and cyclophosphamide (ddAC) alone followed by 4 preoperative cycles of PERJETA in combination with paclitaxel and trastuzumab or trastuzumab hyaluronidase-oysk as given in BERENICE
Following surgery, patients should continue to receive PERJETA and trastuzumab or trastuzumab hyaluronidase-oysk to complete 1 year of treatment (up to 18 cycles).
Adjuvant Treatment Of Breast Cancer
PERJETA should be administered in combination with trastuzumab or trastuzumab hyaluronidase-oysk every 3 weeks for a total of 1 year (up to 18 cycles) or until disease recurrence or unmanageable toxicity, whichever occurs first, as part of a complete regimen for early breast cancer, including standard anthracycline- and/or taxane-based chemotherapy as given in APHINITY. PERJETA and trastuzumab or trastuzumab hyaluronidase-oysk should start on Day 1 of the first taxane-containing cycle [see Clinical Studies].
Dose Modification
For recommendations on delayed or missed doses, please refer to Table 1.
Table 1 : Recommendations regarding delayed or missed doses
| Time between two sequential doses | PERJETA | Trastuzumab (intravenous) | Trastuzumab hyaluronidase-oysk |
| < 6 weeks | Administer PERJETA 420 mg intravenously as soon as possible. Do not wait until the next planned dose. | Administer trastuzumab 6 mg/kg intravenously as soon as possible. Do not wait until the next planned dose. | Administer trastuzumab hyaluronidase-oysk 600 mg/10,000 units subcutaneously as soon as possible. Do not wait until the next planned dose. |
| ≥ 6 weeks | Readminister PERJETA loading dose of 840 mg intravenously as a 60 minute infusion, followed by a maintenance dose of 420 mg administered intravenously over a period of 30 to 60 minutes every 3 weeks thereafter. | Readminister trastuzumab loading dose of 8 mg/kg intravenously over approximately 90 minutes, followed by a maintenance dose of 6 mg/kg administered intraveno usly over a period of 30 or 90 minutes every 3 weeks thereafter. |
PERJETA should be discontinued if trastuzumab or trastuzumab hyaluronidase-oysk treatment is discontinued.
Dose reductions are not recommended for PERJETA.
For chemotherapy dose modifications, see relevant prescribing information.
Left Ventricular Ejection Fraction (LVEF)
Assess left ventricular ejection fraction (LVEF) prior to initiation of PERJETA and at regular intervals during treatment as indicated in Table 2. The recommendations on dose modifications in the event of LVEF dysfunction are also indicated in Table 2 [see WARNINGS AND PRECAUTIONS].
Table 2 : Dose Modifications for Left Ventricular Dysfunction
| Pre- treatment LVEF: | Monitor LVEF every: | Withhold PERJETA and trastuzumab or trastuzumab hyaluronidase-oysk for at least 3 weeks for an LVEF decrease to: | Resume PERJETA and trastuzumab or trastuzumab hyaluronidase-oysk after 3 weeks if LVEF has recovered to: | |||
| Metastatic | ≥ 50% | ~12 weeks | Either | Either | ||
| Breast Cancer | <40% | 40%-45% with a fall of ≥10%-points below pretreatment value | >45% | 40%-45% with a fall of <10%-points below pretreatment value | ||
| Early Breast Cancer | ≥55%* | ~12 weeks (once during neoadjuvant therapy) | <50% with a fall of ≥10%-points below pretreatment value | Either | ||
| ≥50% | <10% points below pretreatment value | |||||
| *For patients receiving anthracycline-based chemotherapy, a LVEF of ≥ 50% is required after completion of anthracyclines, before starting PERJETA and trastuzumab or trastuzumab hyaluronidase-oysk. | ||||||
Infusion-Related Reactions
The infusion rate of PERJETA may be slowed or interrupted if the patient develops an infusion-related reaction [see WARNINGS AND PRECAUTIONS].
Hypersensitivity Reactions/Anaphylaxis
The infusion should be discontinued immediately if the patient experiences a serious hypersensitivity reaction [see WARNINGS AND PRECAUTIONS].
Preparation For Administration
Administer as an intravenous infusion only. Do not administer as an intravenous push or bolus.
Do not mix PERJETA with other drugs.
Preparation
Prepare the solution for infusion, using aseptic technique, as follows:
- Parenteral drug products should be inspected visually for particulates and discoloration prior to administration.
- Withdraw the appropriate volume of PERJETA solution from the vial(s) using a sterile needle and syringe.
- Dilute into a 250 mL 0.9% sodium chloride PVC or non-PVC polyolefin infusion bag.
- Mix diluted solution by gentle inversion. Do not shake.
- Administer immediately once prepared.
- If the diluted infusion solution is not used immediately, it can be stored at 2°C to 8°C for up to 24 hours.
- Dilute with 0.9% Sodium Chloride injection only. Do not use dextrose (5%) solution.
HOW SUPPLIED
Dosage Forms And Strengths
Injection: 420 mg/14 mL (30 mg/mL) in a single-dose vial
Storage And Handling
PERJETA injection is supplied as a 420 mg/14 mL (30 mg/mL) single-dose vial containing preservative-free solution. NDC 50242-145-01.
Store vials in a refrigerator at 2°C to 8°C (36°F to 46°F) until time of use.
Keep vial in the outer carton in order to protect from light.
DO NOT FREEZE. DO NOT SHAKE.
Manufactured by: Genentech, Inc., A Member of the Roche Group, 1 DNA Way, South San Francisco, CA 94080-4990. Revised: Feb 2021
Side Effects for Perjeta
The following adverse reactions are discussed in greater detail in other sections of the label:
- Left Ventricular Dysfunction [see WARNINGS AND PRECAUTIONS]
- Embryo-Fetal Toxicity [see WARNINGS AND PRECAUTIONS]
- Infusion-Related Reactions [see WARNINGS AND PRECAUTIONS]
- Hypersensitivity Reactions/Anaphylaxis [see WARNINGS AND PRECAUTIONS]
Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
Metastatic Breast Cancer (MBC)
The adverse reactions described in Table 3 were identified in 804 patients with HER2-positive metastatic breast cancer treated in CLEOPATRA. Patients were randomized to receive either PERJETA in combination with trastuzumab and docetaxel or placebo in combination with trastuzumab and docetaxel. The median duration of study treatment was 18.1 months for patients in the PERJETA-treated group and 11.8 months for patients in the placebo-treated group. No dose adjustment was permitted for PERJETA or trastuzumab. Adverse reactions resulting in permanent discontinuation of all study therapy were 6% in the PERJETA-treated group and 5% for patients in the placebo-treated group. The most common adverse reactions ( >1%) that led to discontinuation of all study therapy was left ventricular dysfunction (1% for patients in the PERJETA-treated group and 2% for patients in the placebo-treated group). The most common adverse reactions that led to discontinuation of docetaxel alone were edema, fatigue, edema peripheral, neuropathy peripheral, neutropenia, nail disorder and pleural effusion. Table 3 reports the adverse reactions that occurred in at least 10% of patients in the PERJETAtreated group. The safety profile of PERJETA remained unchanged with an additional 2.75 years of follow-up (median total follow-up of 50 months) in CLEOPATRA.
The most common adverse reactions (> 30%) seen with PERJETA in combination with trastuzumab and docetaxel were diarrhea, alopecia, neutropenia, nausea, fatigue, rash, and peripheral neuropathy. The most common NCI - CTCAE v3.0 Grade 3 - 4 adverse reactions (> 2%) were neutropenia, febrile neutropenia, leukopenia, diarrhea, peripheral neuropathy, anemia, asthenia, and fatigue. An increased incidence of febrile neutropenia was observed for Asian patients in both treatment arms compared with patients of other races and from other geographic regions. Among Asian patients, the incidence of febrile neutropenia was higher in the pertuzumab-treated group (26%) compared with the placebo-treated group (12%).
Table 3 : Summary of Adverse Reactions Occurring in ≥ 10% of Patients on the PERJETA Treatment Arm in CLEOPATRA
| Body System/ Adverse Reactions | PERJETA + trastuzumab + docetaxel n=407 Frequency rate % |
Placebo + trastuzumab + docetaxel n=397 Frequency rate % |
||
| All Grades % | Grades 3 - 4 % | All Grades % | Grades 3 - 4 % | |
| General disorders and administration site conditions | ||||
| Fatigue | 37 | 2 | 37 | 3 |
| Mucosal inflammation | 28 | 1 | 20 | 1 |
| Asthenia | 26 | 2 | 30 | 2 |
| Edema peripheral | 23 | 0.5 | 30 | 0.8 |
| Pyrexia | 19 | 1 | 18 | 0.5 |
| Skin and subcutaneous tissue disorders | ||||
| Alopecia | 61 | 0 | 60 | 0.3 |
| Rash | 34 | 0.7 | 24 | 0.8 |
| Nail disorder | 23 | 1 | 23 | 0.3 |
| Pruritus | 14 | 0 | 10 | 0 |
| Dry skin | 11 | 0 | 4 | 0 |
| Gastrointestinal disorders | ||||
| Diarrhea | 67 | 8 | 46 | 5 |
| Nausea | 42 | 1 | 42 | 0.5 |
| Vomiting | 24 | 1 | 24 | 2 |
| Stomatitis | 19 | 0.5 | 15 | 0.3 |
| Constipation | 15 | 0 | 25 | 1 |
| Blood and lymphatic system disorders | ||||
| Neutropenia | 53 | 49 | 50 | 46 |
| Anemia | 23 | 2 | 19 | 4 |
| Leukopenia | 18 | 12 | 20 | 15 |
| Febrile neutropenia* | 14 | 13 | 8 | 7 |
| Nervous system disorders | ||||
| Neuropathy peripheral | 32 | 3 | 34 | 2 |
| Headache | 21 | 1 | 17 | 0.5 |
| Dysgeusia | 18 | 0 | 16 | 0 |
| Dizziness | 13 | 0.5 | 12 | 0 |
| Musculoskeletal and connective tissue disorders | ||||
| Myalgia | 23 | 1 | 24 | 0.8 |
| Arthralgia | 15 | 0.2 | 16 | 0.8 |
| Infections and infestations | ||||
| Upper respiratory tract infection | 17 | 0.7 | 13 | 0 |
| Nasopharyngitis | 12 | 0 | 13 | 0.3 |
| Respiratory, thoracic, and mediastinal disorders | ||||
| Dyspnea | 14 | 1 | 16 | 2 |
| Metabolism and nutrition disorders | ||||
| Decreased appetite | 29 | 2 | 26 | 2 |
| Eye disorders | ||||
| Lacrimation increased | 14 | 0 | 14 | 0 |
| Psychiatric disorders | ||||
| Insomnia | 13 | 0 | 13 | 0 |
| * In this table this denotes an adverse reaction that has been reported in association with a fatal outcome | ||||
The following clinically relevant adverse reactions were reported in < 10% of patients in the PERJETA-treated group in CLEOPATRA:
Infections and infestations: Paronychia (7% in the PERJETA-treated group vs. 4% in the placebo-treated group)
Adverse Reactions Reported In Patients Receiving PERJETA And Trastuzumab After Discontinuation Of Docetaxel
In CLEOPATRA, adverse reactions were reported less frequently after discontinuation of docetaxel treatment. All adverse reactions in the PERJETA and trastuzumab treatment group occurred in < 10% of patients with the exception of diarrhea (19%), upper respiratory tract infection (13%), rash (12%), headache (11%), and fatigue (11%).
Neoadjuvant Treatment Of Breast Cancer (NeoSphere)
In NeoSphere, the most common adverse reactions seen with PERJETA in combination with trastuzumab and docetaxel administered for 4 cycles were similar to those seen in the PERJETAtreated group in CLEOPATRA. The most common adverse reactions ( > 30%) were alopecia, neutropenia, diarrhea, and nausea. The most common NCI - CTCAE v3.0 Grade 3 - 4 adverse reactions ( > 2%) were neutropenia, febrile neutropenia, leukopenia, and diarrhea. In this group, one patient permanently discontinued neoadjuvant treatment due to an adverse event. Table 4 reports the adverse reactions that occurred in patients who received neoadjuvant treatment with PERJETA for breast cancer in NeoSphere.
Table 4 : Summary of Adverse Reactions Occurring in ≥ 10% in the Neoadjuvant Setting for Patients Receiving PERJETA in NeoSphere
| Body System/ Adverse Reactions | Trastuzumab + docetaxel n=107 Frequency rate % |
PERJETA + trastuzumab + docetaxel n=107 Frequency rate % |
PERJETA + trastuzumab n=108 Frequency rate % |
PERJETA + docetaxel n=108 Frequency rate % |
||||
| All Grades % | Grades 3 - 4 % | All Grades % | Grades 3 - 4 % | All Grades % | Grades 3 - 4 % | All Grades % | Grades 3 - 4 % | |
| General disorders and administration site conditions | ||||||||
| Fatigue | 27 | 0 | 26 | 0.9 | 12 | 0 | 26 | 1 |
| Mucosal inflammation | 21 | 0 | 26 | 2 | 3 | 0 | 26 | 0 |
| Asthenia | 18 | 0 | 21 | 2 | 3 | 0 | 16 | 2 |
| Pyrexia | 10 | 0 | 17 | 0 | 8 | 0 | 9 | 0 |
| Edema peripheral | 10 | 0 | 3 | 0 | 0.9 | 0 | 5 | 0 |
| Skin and subcutaneous tissue disorders | ||||||||
| Alopecia | 66 | 0 | 65 | 0 | 3 | 0 | 67 | 0 |
| Rash | 21 | 2 | 26 | 0.9 | 11 | 0 | 29 | 1 |
| Gastrointestinal disorders | ||||||||
| Diarrhea | 34 | 4 | 46 | 6 | 28 | 0 | 54 | 4 |
| Nausea | 36 | 0 | 39 | 0 | 14 | 0 | 36 | 1 |
| Stomatitis | 7 | 0 | 18 | 0 | 5 | 0 | 10 | 0 |
| Vomiting | 12 | 0 | 13 | 0 | 5 | 0 | 16 | 2 |
| Blood and lymphatic system disorders | ||||||||
| Neutropenia | 64 | 59 | 50 | 45 | 0.9 | 0.9 | 65 | 57 |
| Leukopenia | 21 | 11 | 9 | 5 | 0 | 0 | 14 | 9 |
| Nervous system disorders | ||||||||
| Dysgeusia | 10 | 0 | 15 | 0 | 5 | 0 | 7 | 0 |
| Headache | 11 | 0 | 11 | 0 | 14 | 0 | 13 | 0 |
| Peripheral Sensory Neuropathy | 12 | 0.9 | 8 | 0.9 | 2 | 0 | 11 | 0 |
| Musculoskeletal and connective tissue disorders | ||||||||
| Myalgia | 22 | 0 | 22 | 0 | 9 | 0 | 21 | 0 |
| Arthralgia | 8 | 0 | 10 | 0 | 5 | 0 | 10 | 0 |
| Metabolism and nutrition disorders | ||||||||
| Decreased appetite | 7 | 0 | 14 | 0 | 2 | 0 | 15 | 0 |
| Psychiatric disorders | ||||||||
| Insomnia | 11 | 0 | 8 | 0 | 4 | 0 | 9 | 0 |
The following adverse reactions were reported in < 10% of patients receiving neoadjuvant treatment and occurred more frequently in PERJETA-treated groups in NeoSphere: (Ptz=pertuzumab; H=trastuzumab; D=docetaxel)
Blood and lymphatic system disorders: Anemia (7% in the H+D arm, 3% in the Ptz+H+D arm, 5% in the Ptz+H arm and 9% in the Ptz+D arm), Febrile neutropenia (7% in the H+D arm, 8% in the Ptz+H+D arm, 0% in the Ptz+H arm and 7% in the Ptz+D arm)
Nervous system disorders: Dizziness (4% in the H+D arm, 3% in the Ptz+H+D arm, 6% in the Ptz+H arm and 3% in the Ptz+D arm)
Infections and infestations: Upper respiratory tract infection (3% in the H+D arm, 5% in the Ptz+H+D arm, 2% in the Ptz+H arm and 7% in the Ptz+D arm)
Eye disorders: Lacrimation increased (2% in the H+D arm, 4% in the Ptz+H+D arm, 0.9% in the Ptz+H arm, and 4% in the Ptz+D arm)
Neoadjuvant Treatment Of Breast Cancer (TRYPHAENA)
In TRYPHAENA, when PERJETA was administered in combination with trastuzumab and docetaxel for 3 cycles following 3 cycles of FEC, the most common adverse reactions ( > 30%) were diarrhea, nausea, alopecia, neutropenia, vomiting, and fatigue. The most common NCICTCAE (version 3) Grade 3 - 4 adverse reactions ( > 2%) were neutropenia, leukopenia, febrile neutropenia, diarrhea, left ventricular dysfunction, anemia, dyspnea, nausea, and vomiting.
Similarly, when PERJETA was administered in combination with docetaxel, carboplatin, and trastuzumab (TCH) for 6 cycles, the most common adverse reactions ( > 30%) were diarrhea, alopecia, neutropenia, nausea, fatigue, vomiting, anemia, and thrombocytopenia. The most common NCI-CTCAE (version 3) Grade 3 - 4 adverse reactions ( > 2%) were neutropenia, febrile neutropenia, anemia, leukopenia, diarrhea, thrombocytopenia, vomiting, fatigue, ALT increased, hypokalemia, and hypersensitivity.
Adverse reactions resulting in permanent discontinuation of any component of neoadjuvant treatment occurred in 7% of patients receiving PERJETA in combination with trastuzumab and docetaxel following FEC, and 8% for patients receiving PERJETA in combination with TCH. The most common adverse reactions ( >2%) resulting in permanent discontinuation of PERJETA were left ventricular dysfunction, drug hypersensitivity, and neutropenia. Table 5 reports the adverse reactions that occurred in patients who received neoadjuvant treatment with PERJETA for breast cancer in TRYPHAENA.
Table 5 : Summary of Adverse Reactions Occurring in ≥ 10% of Patients Receiving Neoadjuvant Treatment with PERJETA in TRYPHAENA
| Body System/Adverse Reactions | PERJETA + trastuzumab + FEC followed by PERJETA + trastuzumab + docetaxel n=72 Frequency rate % |
PERJETA + trastuzumab + docetaxel following FEC n=75 Frequency rate % |
PERJETA + TCH n=76 Frequency rate % |
|||
| All Grades % | Grades 3 - 4 % | All Grades % | Grades 3 - 4 % | All Grades % | Grades 3 - 4 % | |
| General disorders and administration site conditions | ||||||
| Fatigue | 36 | 0 | 36 | 0 | 42 | 4 |
| Mucosal inflammation | 24 | 0 | 20 | 0 | 17 | 1 |
| Pyrexia | 17 | 0 | 9 | 0 | 16 | 0 |
| Asthenia | 10 | 0 | 15 | 1 | 13 | 1 |
| Edema peripheral | 11 | 0 | 4 | 0 | 9 | 0 |
| Skin and subcutaneous tissue disorders | ||||||
| Alopecia | 49 | 0 | 52 | 0 | 55 | 0 |
| Rash | 19 | 0 | 11 | 0 | 21 | 1 |
| Palmar-Plantar Erythrodysaesthesia Syndrome | 7 | 0 | 11 | 0 | 8 | 0 |
| Dry skin | 6 | 0 | 9 | 0 | 11 | 0 |
| Gastrointestinal disorders | ||||||
| Diarrhea | 61 | 4 | 61 | 5 | 72 | 12 |
| Nausea | 53 | 0 | 53 | 3 | 45 | 0 |
| Vomiting | 40 | 0 | 36 | 3 | 39 | 5 |
| Dyspepsia | 25 | 1 | 8 | 0 | 22 | 0 |
| Constipation | 18 | 0 | 23 | 0 | 16 | 0 |
| Stomatitis | 14 | 0 | 17 | 0 | 12 | 0 |
| Blood and lymphatic system disorders | ||||||
| Neutropenia | 51 | 47 | 47 | 43 | 49 | 46 |
| Leukopenia | 22 | 19 | 16 | 12 | 17 | 12 |
| Anemia | 19 | 1 | 9 | 4 | 38 | 17 |
| Febrile neutropenia | 18 | 18 | 9 | 9 | 17 | 17 |
| Thrombocytopenia | 7 | 0 | 1 | 0 | 30 | 12 |
| Immune system disorders | ||||||
| Hypersensitivity | 10 | 3 | 1 | 0 | 12 | 3 |
| Nervous system disorders | ||||||
| Headache | 22 | 0 | 15 | 0 | 17 | 0 |
| Dysgeusia | 11 | 0 | 13 | 0 | 21 | 0 |
| Dizziness | 8 | 0 | 8 | 1 | 16 | 0 |
| Neuropathy peripheral | 6 | 0 | 1 | 0 | 11 | 0 |
| Musculoskeletal and connective tissue disorders | ||||||
| Myalgia | 17 | 0 | 11 | 1 | 11 | 0 |
| Arthralgia | 11 | 0 | 12 | 0 | 7 | 0 |
| Respiratory, thoracic, and mediastinal disorders | ||||||
| Dyspnea | 13 | 0 | 8 | 3 | 11 | 1 |
| Epistaxis | 11 | 0 | 11 | 0 | 16 | 1 |
| Cough | 10 | 0 | 5 | 0 | 12 | 0 |
| Oropharyngeal pain | 8 | 0 | 7 | 0 | 12 | 0 |
| Metabolism and nutrition disorders | ||||||
| Decreased appetite | 21 | 0 | 11 | 0 | 21 | 0 |
| Eye disorders | ||||||
| Lacrimation increased | 13 | 0 | 5 | 0 | 8 | 0 |
| Psychiatric disorders | ||||||
| Insomnia | 11 | 0 | 13 | 0 | 21 | 0 |
| Investigations | ||||||
| ALT increased | 7 | 0 | 3 | 0 | 11 | 4 |
| FEC=5-fluorouracil, epirubicin, cyclophosphamide, TCH=docetaxel, carboplatin, trastuzumab | ||||||
The following selected adverse reactions were reported in < 10% of patients receiving neoadjuvant treatment in TRYPHAENA: (Ptz=pertuzumab; H=trastuzumab; D=docetaxel; FEC= fluorouracil, epirubicin, and cyclophosphamide; TCH=docetaxel, carboplatin, and trastuzumab)
Skin and subcutaneous tissue disorders: Nail disorder (10% in the Ptz+H+FEC/Ptz+H+D arm, 7% in the FEC/Ptz+H+D arm, and 9% in the Ptz+TCH arm), Paronychia (0% in the Ptz+H+FEC/Ptz+H+D arm, and 1% in both the FEC/Ptz+H+D and Ptz+TCH arms), Pruritus (3% in the Ptz+H+FEC/Ptz+H+D arm, 4% in the FEC/Ptz+H+D arm, and 4% in the Ptz+TCH arm)
Infections and infestations: Upper respiratory tract infection (8.3% in the Ptz+H+FEC/Ptz+H+D arm, 4.0% in the FEC/Ptz+H+D arm, and 2.6% in the Ptz+TCH arm), Nasopharyngitis (6.9% in the Ptz+H+FEC/Ptz+H+D arm, 6.7% in the FEC/Ptz+H+D arm, and 7.9% in the Ptz+TCH arm)
Neoadjuvant Treatment Of Breast Cancer (BERENICE)
In BERENICE, when PERJETA was administered in combination with trastuzumab and paclitaxel for 4 cycles following 4 cycles of ddAC, the most common adverse reactions ( > 30%) were nausea, diarrhea, alopecia, fatigue, constipation, peripheral neuropathy and headache. The most common Grade 3 - 4 adverse reactions ( > 2%) were neutropenia, febrile neutropenia, neutrophil count decreased, white blood cell count decreased, anemia, diarrhea, peripheral neuropathy, alanine aminotransferase increased and nausea.
When PERJETA was administered in combination with trastuzumab and docetaxel for 4 cycles following 4 cycles of FEC, the most common adverse reactions ( > 30%) were diarrhea, nausea, alopecia, asthenia, constipation, fatigue, mucosal inflammation, vomiting, myalgia, and anemia. The most common Grade 3 - 4 adverse reactions ( > 2%) were febrile neutropenia, diarrhea, neutropenia, neutrophil count decreased, stomatitis, fatigue, vomiting, mucosal inflammation, neutropenic sepsis and anemia.
Adverse reactions resulting in permanent discontinuation of any component of neoadjuvant treatment were 14% for patients receiving PERJETA in combination with trastuzumab and paclitaxel following ddAC and 8% for patients receiving PERJETA in combination with trastuzumab and docetaxel following FEC. The most common adverse reactions ( >1%) resulting in permanent discontinuation of any component of neoadjuvant treatment were neuropathy peripheral, ejection fraction decreased, diarrhea, neutropenia and infusion related reaction. Table 6 reports the adverse reactions that occurred in patients who received neoadjuvant treatment with PERJETA for breast cancer in BERENICE.
Table 6 : Summary of Adverse Reactions Occurring in ≥ 10% of Patients Receiving Neoadjuvant Treatment with PERJETA in BERENICE
| Body System/Adverse Reactions | PERJETA + trastuzumab + paclitaxel following ddAC n=199 Frequency rate % |
PERJETA + trastuzumab + docetaxel following FEC n=198 Frequency rate % |
||
| All Grades % | Grades 3 - 4 % | All Grades % | Grades 3 - 4 % | |
| General disorders and administration site conditions | ||||
| Fatigue | 58 | 1 | 38 | 5 |
| Asthenia | 19 | 2 | 41 | 0 |
| Mucosal inflammation | 22 | 1 | 37 | 4 |
| Pyrexia | 15 | 0 | 18 | 0 |
| Edema peripheral | 9 | 0 | 12 | 1 |
| Skin and subcutaneous tissue disorders | ||||
| Alopecia | 62 | 0 | 59 | 0 |
| Rash | 14 | 0 | 11 | 0 |
| Dry skin | 14 | 0 | 10 | 0 |
| Nail discoloration | 15 | 0 | 2 | 0 |
| Palmar-Plantar Erythrodysaesthesia Syndrome | 6 | 0 | 10 | 0.5 |
| Gastrointestinal disorders | ||||
| Nausea | 71 | 3 | 69 | 2 |
| Diarrhea | 67 | 3 | 69 | 10 |
| Constipation | 35 | 0.5 | 38 | 0.5 |
| Vomiting | 23 | 1 | 35 | 4 |
| Stomatitis | 25 | 0 | 27 | 5 |
| Dyspepsia | 19 | 0 | 16 | 0 |
| Abdominal pain upper | 6 | 0 | 13 | 0 |
| Abdominal pain | 5 | 0 | 10 | 0 |
| Gastroesophageal reflux disease | 12 | 0 | 2 | 0 |
| Blood and lymphatic system disorders | ||||
| Anemia | 27 | 3 | 30 | 3 |
| Neutropenia | 22 | 12 | 16 | 9 |
| Febrile neutropenia | 7 | 7 | 17 | 17 |
| Nervous system disorders | ||||
| Headache | 30 | 0.5 | 14 | 0.5 |
| Dysgeusia | 20 | 0 | 19 | 0.5 |
| Neuropathy peripheral | 42 | 3 | 26 | 0.5 |
| Paresthesia | 15 | 0 | 9 | 0 |
| Dizziness | 12 | 0 | 8 | 0 |
| Musculoskeletal and connective tissue disorders | ||||
| Myalgia | 20 | 0 | 33 | 1 |
| Arthralgia | 20 | 0 | 21 | 1 |
| Back pain | 10 | 0 | 9 | 0 |
| Pain in extremity | 10 | 0 | 8 | 0 |
| Bone pain | 12 | 0.5 | 5 | 0 |
| Infections and infestations | ||||
| Urinary tract infection | 11 | 1 | 2 | 0 |
| Respiratory, thoracic, and mediastinal disorders | ||||
| Epistaxis | 25 | 0 | 19 | 0 |
| Dyspnea | 15 | 0.5 | 15 | 0.5 |
| Cough | 20 | 0.5 | 9 | 0 |
| Oropharyngeal pain | 10 | 0 | 8 | 0.5 |
| Metabolism and nutrition disorders | ||||
| Decreased appetite | 20 | 0 | 23 | 0 |
| Eye disorders | ||||
| Lacrimation increased | 9 | 0 | 18 | 0 |
| Psychiatric disorders | ||||
| Insomnia | 19 | 0 | 13 | 0 |
| Vascular disorders | ||||
| Hot flush | 19 | 0 | 13 | 0 |
| Investigations | ||||
| White blood cell count decreased | 11 | 4 | 3 | 2 |
| Injury, poisoning and procedural complications | ||||
| Infusion related reaction | 16 | 1 | 13 | 1 |
| ddAC = dose-dense doxorubicin, cyclophosphamide, FEC=5-fluorouracil, epirubicin, cyclophosphamide | ||||
The following selected adverse reactions were reported in < 10% of patients receiving neoadjuvant treatment in BERENICE: (Ptz=pertuzumab; H=trastuzumab; P=paclitaxel; ddAC=dose-dense doxorubicin and cyclophosphamide; D=docetaxel; FEC= fluorouracil, epirubicin, and cyclophosphamide)
Skin and Subcutaneous tissue disorders: Pruritus (9% in the ddAC/Ptz+H+P arm, and 8% in the FEC/Ptz+H+D arm), Nail disorder (7% in the ddAC/Ptz+H+P arm, and 10% in the FEC/Ptz+H+D arm)
Infections and infestations: Upper respiratory tract infection (7% in the ddAC/Ptz+H+P arm, and 2% in the FEC/Ptz+H+D arm), nasopharyngitis (7% in the ddAC/Ptz+H+P arm, and 9% in the FEC/Ptz+H+D arm), paronychia (0.5% in the ddAC/Ptz+H+P arm, and 1% in the FEC/Ptz+H+D arm)
Adjuvant Treatment Of Breast Cancer (APHINITY)
The adverse reactions described in Table 7 were identified in 4769 patients with HER2-positive early breast cancer treated in APHINITY. Patients were randomized to receive either PERJETA in combination with trastuzumab and chemotherapy or placebo in combination with trastuzumab and chemotherapy.
Adverse reactions resulting in permanent discontinuation of any study therapy were 13% for patients in the PERJETA-treated group and 12% for patients in the placebo-treated group. Adverse reactions resulting in permanent discontinuation of PERJETA or placebo was 7% and 6%, respectively. The most common adverse reactions ( >0.5%) resulting in permanent discontinuation of any study treatment were ejection fraction decreased, neuropathy peripheral, diarrhea, and cardiac failure. Table 7 reports the adverse reactions that occurred in at least 10% of patients in the PERJETA-treated group.
When PERJETA was administered in combination with trastuzumab and chemotherapy, the most common adverse reactions ( > 30%) were diarrhea, nausea, alopecia, fatigue, peripheral neuropathy, and vomiting. The most common Grade 3 - 4 adverse reactions ( > 2%) were neutropenia, febrile neutropenia, diarrhea, neutrophil count decreased, anemia, white blood cell count decreased, leukopenia, fatigue, nausea, and stomatitis.
The incidence of diarrhea, all Grades, was higher when chemotherapy was administered with targeted therapy (61% in the PERJETA-treated group vs. 34% in the placebo-treated group), and was higher when administered with non-anthracycline based therapy (85% in the PERJETAtreated group vs. 62% in the placebo-treated group) than with anthracycline based therapy (67% in the PERJETA-treated group vs. 41% in the placebo-treated group). The incidence of diarrhea during the period that targeted therapy was administered without chemotherapy was 18% in the PERJETA-treated group vs. 9% in the placebo-treated group. The median duration of all Grades diarrhea was 8 days for the PERJETA-treated group vs. 6 days for the placebo-treated group. The median duration of Grade ≥3 diarrhea was 20 days for the PERJETA-treated group vs. 8 days for the placebo-treated group. More patients required hospitalization for diarrhea as a serious adverse event in the PERJETA-treated group (2.4%) than in the placebo-treated group (0.7%).
Table 7 : Summary of Adverse Reactions Occurring in ≥ 10% of Patients Receiving Adjuvant Treatment with PERJETA in APHINITY
| Body System/ Adverse Reactions | PERJETA + trastuzumab + chemotherapy n=2364 Frequency rate % |
Placebo + trastuzumab + chemotherapy n=2405 Frequency rate % |
||
| All Grades % | Grades 3 - 4 % | All Grades % | Grades 3 - 4 % | |
| General disorders and administration site conditions | ||||
| F atigue | 49 | 4 | 44 | 3 |
| Mucosal inflammation | 23 | 2 | 19 | 0.7 |
| Asthenia | 21 | 1 | 21 | 2 |
| Pyrexia | 20 | 0.6 | 20 | 0.7 |
| Edema peripheral | 17 | 0 | 20 | 0.2 |
| Skin and subcutaneous tissue disorders | ||||
| Alopecia | 67 | <0.1 | 67 | <0.1 |
| Rash | 26 | 0.4 | 20 | 0.2 |
| Pruritus | 14 | 0.1 | 9 | <0.1 |
| Dry skin | 13 | 0.1 | 11 | <0.1 |
| Nail disorder | 12 | 0.2 | 12 | 0.1 |
| Gastrointestinal disorders | ||||
| Diarrhea | 71 | 10 | 45 | 4 |
| Nausea | 69 | 2 | 65 | 2 |
| Vomiting | 32 | 2 | 30 | 2 |
| Constipation | 29 | 0.5 | 32 | 0.3 |
| Stomatitis | 28 | 2 | 24 | 1 |
| Dyspepsia | 14 | 0 | 14 | 0 |
| Abdominal pain | 12 | 0.5 | 11 | 0.6 |
| Abdominal pain upper | 10 | 0.3 | 9 | 0.2 |
| Blood and lymphatic system disorders | ||||
| Anemia | 28 | 7 | 23 | 5 |
| Neutropenia | 25 | 16 | 23 | 16 |
| Febrile neutropenia* | 12 | 12 | 11 | 11 |
| Nervous system disorders | ||||
| Dysgeusia | 26 | 0.1 | 22 | <0.1 |
| Neuropathy peripheral | 33 | 1 | 32 | 1 |
| Headache | 22 | 0.3 | 23 | 0.4 |
| Paresthesia | 12 | 0.5 | 10 | 0.2 |
| Dizziness | 11 | 0 | 11 | 0.2 |
| Musculoskeletal and connective tissue disorders | ||||
| Arthralgia | 29 | 0.9 | 33 | 1 |
| Myalgia | 26 | 0.9 | 30 | 1 |
| Pain in extremity | 10 | 0.2 | 10 | 0.2 |
| Infections and infestations | ||||
| Nasopharyngitis | 13 | <0.1 | 12 | 0.1 |
| Respiratory, thoracic, and mediastinal disorders | ||||
| Epistaxis | 18 | <0.1 | 14 | 0 |
| Cough | 16 | <0.1 | 15 | <0.1 |
| Dyspnea | 12 | 0.4 | 12 | 0.5 |
| Metabolism and nutrition disorders | ||||
| Decreased appetite | 24 | 0.8 | 20 | 0.4 |
| Vascular disorders | ||||
| Hot flush | 20 | 0.2 | 21 | 0.4 |
| Eye disorders | ||||
| Lacrimation increased | 13 | 0 | 13 | <0.1 |
| Psychiatric disorders | ||||
| Insomnia | 17 | 0.3 | 17 | <0.1 |
| Investigations | ||||
| Neutrophil count decreased | 14 | 10 | 14 | 10 |
| Injury, poisoning and procedural complications | ||||
| Radiation skin injury | 13 | 0.3 | 11 | 0.3 |
| * In this table this denotes an adverse reaction that has been reported in association with a fatal outcome | ||||
For the adverse reactions that were reported in ≥10% of patients with at least 5% difference between the PERJETA-treated group and the placebo-treated group in APHINITY, the breakdown per chemotherapy regimen is provided: (Ptz=pertuzumab; H=trastuzumab; AC=anthracyclines; TCH=docetaxel, carboplatin, and trastuzumab)
Gastrointestinal disorders: Diarrhea (67% in the Ptz+H+AC chemo arm, 85% in the Ptz+TCH arm, 41% in the Pla+H+AC chemo arm, 62% in the Pla+TCH arm)
Skin and subcutaneous disorders: Rash (26% in the Ptz+H+AC chemo arm, 25% in the Ptz+TCH arm, 21% in the Pla+H+AC chemo arm, 19% in the Pla+TCH arm), Pruritus (14% in the Ptz+H+AC chemo arm, 15% in the Ptz+TCH arm, 9% in the Pla+H+AC chemo arm, 9% in the Pla+TCH arm)
The following clinically relevant adverse reactions were reported in < 10% of patients in the PERJETA-treated group in APHINITY:
Blood and lymphatic system disorders: Leukopenia (9% in the PERJETA-treated group vs. 9% in the placebo-treated group)
Infections and infestations: Upper respiratory tract infection (8% in the PERJETA-treated group vs. 7% in the placebo-treated group), paronychia (4% in the PERJETA-treated group vs. 2% in the placebo-treated group)
Adverse Reactions Reported In Patients Receiving PERJETA And Trastuzumab After Discontinuation Of Chemotherapy
In the APHINITY study, during the targeted treatment alone phase, all adverse reactions in the PERJETA treatment group occurred in < 10% of patients with the exception of diarrhea (18%), arthralgia (15%), radiation skin injury (12%), and hot flush (12%).
Immunogenicity
As with all therapeutic proteins, there is the potential for immunogenicity. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to pertuzumab in the studies described below with the incidence of antibodies in other studies or to other products may be misleading.
Patients in CLEOPATRA were tested at multiple time-points for antibodies to PERJETA. 3% (13/389) of patients in the PERJETA-treated group and 7% (25/372) of patients in the placebotreated group tested positive for anti-PERJETA antibodies. Of these 38 patients, none experienced anaphylactic/hypersensitivity reactions that were clearly related to the anti-drug antibodies (ADA). The presence of pertuzumab in patient serum at the levels expected at the time of ADA sampling can interfere with the ability of this assay to detect anti-pertuzumab antibodies. In addition, the assay may be detecting antibodies to trastuzumab. As a result, data may not accurately reflect the true incidence of anti-pertuzumab antibody development.
In the neoadjuvant period of BERENICE, 0.3% (1/383) of patients treated with PERJETA tested positive for anti-PERJETA antibodies. This patient did not experience any anaphylactic/hypersensitivity reactions.
Post-Marketing Experience
The following adverse reactions have been identified during post-approval use of PERJETA. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
- Tumor lysis syndrome (TLS): Cases of possible TLS have been reported in patients treated with PERJETA. Patients with significant tumor burden (e.g., bulky metastases) may be at a higher risk. Patients could present with hyperuricemia, hyperphosphatemia, and acute renal failure which may represent possible TLS. Providers should consider additional monitoring and/or treatment as clinically indicated.
Drug Interactions for Perjeta
No drug-drug interactions were observed between pertuzumab and trastuzumab, or between pertuzumab and docetaxel, paclitaxel, or carboplatin.
Warnings for Perjeta
Included as part of the PRECAUTIONS section.
Precautions for Perjeta
Left Ventricular Dysfunction
Decreases in LVEF have been reported with drugs that block HER2 activity, including PERJETA. Assess LVEF prior to initiation of PERJETA and at regular intervals during treatment to ensure that LVEF is within normal limits. If the LVEF declines and has not improved, or has declined further at the subsequent assessment, discontinuation of PERJETA and trastuzumab should be strongly considered [DOSAGE AND ADMINISTRATION].
In CLEOPATRA, for patients with MBC, PERJETA in combination with trastuzumab and docetaxel was not associated with increases in the incidence of symptomatic left ventricular systolic dysfunction (LVSD) or decreases in LVEF compared with placebo in combination with trastuzumab and docetaxel [see Clinical Studies]. Left ventricular dysfunction occurred in 4% of patients in the PERJETA-treated group and 8% of patients in the placebo-treated group. Symptomatic left ventricular systolic dysfunction (congestive heart failure) occurred in 1% of patients in the PERJETA-treated group and 2% of patients in the placebo-treated group [see ADVERSE REACTIONS]. Patients who have received prior anthracyclines or prior radiotherapy to the chest area may be at higher risk of decreased LVEF.
In patients receiving neoadjuvant treatment in NeoSphere, the incidence of LVSD was higher in the PERJETA-treated groups compared to the trastuzumab- and docetaxel-treated group. An increased incidence of LVEF declines was observed in patients treated with PERJETA in combination with trastuzumab and docetaxel. In the overall treatment period, LVEF decline > 10% and a drop to less than 50% occurred in 2% of patients treated with neoadjuvant trastuzumab and docetaxel as compared to 8% of patients treated with neoadjuvant PERJETA in combination with trastuzumab and docetaxel. Left ventricular dysfunction occurred in 0.9% of patients treated with neoadjuvant trastuzumab and docetaxel as compared to 3% of patients treated with neoadjuvant PERJETA in combination with trastuzumab and docetaxel. Symptomatic LVSD occurred in 0.9% of patients treated with neoadjuvant PERJETA in combination with trastuzumab and no patients in the other 3 arms. LVEF recovered to ≥ 50% in all patients.
In patients receiving neoadjuvant PERJETA in TRYPHAENA, in the overall treatment period, LVEF decline > 10% and a drop to less than 50% occurred in 7% of patients treated with PERJETA plus trastuzumab and FEC followed by PERJETA plus trastuzumab and docetaxel, 16% of patients treated with PERJETA plus trastuzumab and docetaxel following FEC, and 11% of patients treated with PERJETA in combination with TCH. Left ventricular dysfunction occurred in 6% of patients treated with PERJETA plus trastuzumab and FEC followed by PERJETA plus trastuzumab and docetaxel, 4% of patients treated with PERJETA plus trastuzumab and docetaxel following FEC, and 3% of patients treated with PERJETA in combination with TCH. Symptomatic LVSD occurred in 4% of patients treated with PERJETA plus trastuzumab and docetaxel following FEC, 1% of patients treated with PERJETA in combination with TCH, and none of the patients treated with PERJETA plus trastuzumab and FEC followed by PERJETA plus trastuzumab and docetaxel. LVEF recovered to ≥ 50% in all but one patient.
In patients receiving neoadjuvant PERJETA in BERENICE, in the neoadjuvant period, LVEF decline ≥ 10% and a drop to less than 50% as measured by ECHO/MUGA assessment occurred in 7% of patients treated with PERJETA plus trastuzumab and paclitaxel following ddAC, and 2% of patients treated with PERJETA plus trastuzumab and docetaxel following FEC. Ejection fraction decreased (asymptomatic LVD) occurred in 7% of patients treated with PERJETA plus trastuzumab and paclitaxel following ddAC and 4% of the patients treated with PERJETA plus trastuzumab and docetaxel following FEC in the neoadjuvant period. Symptomatic LVSD (NYHA Class III/IV Congestive Heart Failure) occurred in 2% of patients treated with PERJETA plus trastuzumab and paclitaxel following ddAC and none of the patients treated with PERJETA plus trastuzumab and docetaxel following FEC in the neoadjuvant period.
In patients receiving adjuvant PERJETA in APHINITY, the incidence of symptomatic heart failure (NYHA Class III/IV) with a LVEF decline ≥ 10% and a drop to less than 50% was <1% (0.6% of PERJETA-treated patients vs. 0.2% of placebo-treated patients). Of the patients who experienced symptomatic heart failure, 47% of PERJETA-treated patients and 67% of placebotreated patients had recovered (defined as 2 consecutive LVEF measurements above 50%) at the data cutoff. The majority of the events (86%) were reported in anthracycline-treated patients. Asymptomatic or mildly symptomatic (NYHA Class II) declines in LVEF ≥ 10% and a drop to less than 50% were reported in 3% of PERJETA-treated patients and 3% of placebo-treated patients, of whom 80% of PERJETA-treated patients and 81% of placebo-treated patients recovered at the data cutoff.
PERJETA has not been studied in patients with a pretreatment LVEF value of < 50%, a prior history of CHF, decreases in LVEF to < 50% during prior trastuzumab therapy, or conditions that could impair left ventricular function such as uncontrolled hypertension, recent myocardial infarction, serious cardiac arrhythmia requiring treatment or a cumulative prior anthracycline exposure to > 360 mg/m² of doxorubicin or its equivalent.
Embryo-Fetal Toxicity
Based on its mechanism of action and findings in animal studies, PERJETA can cause fetal harm when administered to a pregnant woman. PERJETA is a HER2/neu receptor antagonist. Cases of oligohydramnios and oligohydramnios sequence manifesting as pulmonary hypoplasia, skeletal abnormalities, and neonatal death have been reported with use of another HER2/neu receptor antagonist (trastuzumab) during pregnancy. In an animal reproduction study, administration of pertuzumab to pregnant cynomolgus monkeys during the period of organogenesis resulted in oligohydramnios, delayed fetal kidney development, and embryo-fetal death at exposures 2.5 to 20 times the exposure in humans at the recommended dose, based on Cmax.
Verify the pregnancy status of females of reproductive potential prior to the initiation of PERJETA. Advise pregnant women and females of reproductive potential that exposure to PERJETA in combination with trastuzumab during pregnancy or within 7 months prior to conception can result in fetal harm, including embryo-fetal death or birth defects. Advise females of reproductive potential to use effective contraception during treatment and for 7 months following the last dose of PERJETA in combination with trastuzumab [see Use In Specific Populations].
Infusion-Related Reactions
PERJETA has been associated with infusion reactions, including fatal events. [see ADVERSE REACTIONS]. An infusion reaction was defined in CLEOPATRA as any event described as hypersensitivity, anaphylactic reaction, acute infusion reaction, or cytokine release syndrome occurring during an infusion or on the same day as the infusion. The initial dose of PERJETA was given the day before trastuzumab and docetaxel to allow for the examination of PERJETAassociated reactions. On the first day, when only PERJETA was administered, the overall frequency of infusion reactions was 13% in the PERJETA-treated group and 10% in the placebotreated group. Less than 1% were Grade 3 or 4. The most common infusion reactions (≥ 1.0%) were pyrexia, chills, fatigue, headache, asthenia, hypersensitivity, and vomiting.
During the second cycle when all drugs were administered on the same day, the most common infusion reactions in the PERJETA-treated group (≥ 1.0%) were fatigue, dysgeusia, hypersensitivity, myalgia, and vomiting.
In NeoSphere, TRYPHAENA, and APHINITY, PERJETA was administered on the same day as the other study treatment drugs. For APHINITY, infusion-related reactions occurred in 21% of patients on the first day of PERJETA administration (in combination with trastuzumab and chemotherapy) and in 18% of patients in the placebo arm. The incidence of Grade 3-4 National Cancer Institute - Common Terminology Criteria for Adverse Events (NCI - CTCAE v4.0) reactions was 1% for the PERJETA arm and 0.7% for the placebo arm.
Observe patients closely for 60 minutes after the first infusion and for 30 minutes after subsequent infusions of PERJETA. If a significant infusion-related reaction occurs, slow or interrupt the infusion, and administer appropriate medical therapies. Monitor patients carefully until complete resolution of signs and symptoms. Consider permanent discontinuation in patients with severe infusion reactions [see DOSAGE AND ADMINISTRATION].
Hypersensitivity Reactions/Anaphylaxis
In CLEOPATRA, the overall frequency of hypersensitivity/anaphylaxis reactions was 11% in the PERJETA-treated group and 9% in the placebo-treated group. The incidence of Grade 3 – 4 hypersensitivity/anaphylaxis reactions was 2% in the PERJETA-treated group and 3% in the placebo-treated group according to NCI - CTCAE v3.0. Overall, 4 patients in the PERJETAtreated group and 2 patients in the placebo-treated group experienced anaphylaxis.
In NeoSphere, TRYPHAENA, BERENICE, and APHINITY, hypersensitivity/anaphylaxis events were consistent with those observed in CLEOPATRA. In NeoSphere, two patients in the PERJETA- and docetaxel-treated group experienced anaphylaxis. In APHINITY, the overall frequency of hypersensitivity/anaphylaxis was 5% in the PERJETA treated group vs. 4% in the placebo-treated group. The incidence was highest in the PERJETA plus TCH treated group (8%) of which 1% were NCI-CTCAE (v4.0) Grade 3 – 4.
Patients should be observed closely for hypersensitivity reactions. Severe hypersensitivity, including anaphylaxis and fatal events, have been observed in patients treated with PERJETA [see Clinical Trials Experience]. Angioedema has been described in post-marketing reports. Medications to treat such reactions, as well as emergency equipment, should be available for immediate use. PERJETA is contraindicated in patients with known hypersensitivity to pertuzumab or to any of its excipients [see CONTRAINDICATIONS].
Nonclinical Toxicology
Carcinogenesis, Mutagenesis, Impairment Of Fertility
Long-term studies in animals have not been performed to evaluate the carcinogenic potential of pertuzumab.
Studies have not been performed to evaluate the mutagenic potential of pertuzumab.
No specific fertility studies in animals have been performed to evaluate the effect of pertuzumab. No adverse effects on male and female reproductive organs were observed in repeat-dose toxicity studies of up to six months duration in cynomolgus monkeys.
Use In Specific Populations
Pregnancy
Pregnancy Pharmacovigilance Program
There is a pregnancy pharmacovigilance program for PERJETA. If PERJETA is administered during pregnancy, or if a patient becomes pregnant while receiving PERJETA or within 7 months following the last dose of PERJETA in combination with trastuzumab, health care providers and patients should immediately report PERJETA exposure to Genentech at 1-888- 835-2555.
Risk Summary
Based on its mechanism of action and findings in animal studies, PERJETA can cause fetal harm when administered to a pregnant woman. There are no available data on the use of PERJETA in pregnant women. However, in post-marketing reports, use of another HER2/neu receptor antagonist (trastuzumab) during pregnancy resulted in cases of oligohydramnios and oligohydramnios sequence manifesting as pulmonary hypoplasia, skeletal abnormalities, and neonatal death. In an animal reproduction study, administration of pertuzumab to pregnant cynomolgus monkeys during the period of organogenesis resulted in oligohydramnios, delayed fetal kidney development, and embryo-fetal deaths at clinically relevant exposures that were 2.5 to 20-fold greater than exposures in humans receiving the recommended dose, based on Cmax [see Data]. Apprise the patient of the potential risks to a fetus. There are clinical considerations if PERJETA in combination with trastuzumab is used during pregnancy or within 7 months prior to conception [see Clinical Considerations].
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Clinical Considerations
Fetal/Neonatal Adverse Reactions
Monitor women who received PERJETA in combination with trastuzumab during pregnancy or within 7 months prior to conception for oligohydramnios. If oligohydramnios occurs, perform fetal testing that is appropriate for gestational age and consistent with community standards of care.
Data
Animal Data
Pregnant cynomolgus monkeys were treated on Gestational Day (GD)19 with loading doses of 30 to 150 mg/kg pertuzumab, followed by bi-weekly doses of 10 to 100 mg/kg. These dose levels resulted in clinically relevant exposures of 2.5 to 20-fold greater than exposures in humans receiving the recommended dose, based on Cmax. Intravenous administration of pertuzumab from GD19 through GD50 (period of organogenesis) was embryotoxic, with dose-dependent increases in embryo-fetal death between GD25 to GD70. The incidences of embryo-fetal loss were 33, 50, and 85% for dams treated with bi-weekly pertuzumab doses of 10, 30, and 100 mg/kg, respectively (2.5 to 20-fold greater than the recommended human dose, based on Cmax). At Caesarean section on GD100, oligohydramnios, decreased relative lung and kidney weights, and microscopic evidence of renal hypoplasia consistent with delayed renal development were identified in all pertuzumab dose groups. Pertuzumab exposure was reported in offspring from all treated groups, at levels of 29% to 40% of maternal serum levels at GD100.
Lactation
Risk Summary
There is no information regarding the presence of pertuzumab in human milk, the effects on the breastfed infant or the effects on milk production. Published data suggest that human IgG is present in human milk but does not enter the neonatal and infant circulation in substantial amounts. Consider the developmental and health benefits of breast feeding along with the mother’s clinical need for PERJETA treatment and any potential adverse effects on the breastfed child from PERJETA or from the underlying maternal condition. This consideration should also take into account the elimination half-life of pertuzumab and the trastuzumab wash out period of 7 months.
Females And Males Of Reproductive Potential
Pregnancy Testing
Verify the pregnancy status of females of reproductive potential prior to the initiation of PERJETA.
Contraception
Females
Based on the mechanism of action and animal data, PERJETA can cause embryo-fetal harm when administered during pregnancy. Advise females of reproductive potential to use effective contraception during treatment and for 7 months following the last dose of PERJETA in combination with trastuzumab [see Use In Specific Populations].
Pediatric Use
The safety and effectiveness of PERJETA have not been established in pediatric patients.
Geriatric Use
In studies in the indicated populations, CLEOPATRA, NeoSphere, TRYPHAENA, BERENICE, and APHINITY, 464 patients who received PERJETA were ≥ 65 years of age and 47 were ≥ 75 years of age. The most common (≥ 10%) Grade 3-4 adverse reactions in both age groups were neutropenia (22% ≥ 65 years, 23% ≥ 75 years), febrile neutropenia (12% ≥ 65 years, 13% ≥ 75 years), diarrhea (15% ≥ 65 years, 17% ≥ 75 years) and anemia (15% ≥ 75 years). The incidence of the following all grade adverse events was at least 5% higher in patients aged ≥ 65 years of age, compared to patients aged < 65 years of age: decreased appetite (13% higher), anemia (7% higher), weight decreased (7% higher), asthenia (7% higher), dysgeusia (7% higher), neuropathy peripheral and hypomagnesemia (both 5% higher).
No overall differences in efficacy of PERJETA were observed in patients aged ≥ 65 and <65 years of age. There are too few patients aged ≥ 75 years to draw conclusions on efficacy in this age group.
Based on a population pharmacokinetic analysis, no significant difference was observed in the pharmacokinetics of pertuzumab between patients < 65 years (n=306) and patients ≥ 65 years (n=175).
Renal Impairment
Dose adjustments of PERJETA are not needed in patients with mild (creatinine clearance [CLcr] 60 to 90 mL/min) or moderate (CLcr 30 to 60 mL/min) renal impairment. No dose adjustment can be recommended for patients with severe renal impairment (CLcr less than 30 mL/min) because of the limited pharmacokinetic data available [see CLINICAL PHARMACOLOGY].
Hepatic Impairment
No clinical studies have been conducted to evaluate the effect of hepatic impairment on the pharmacokinetics of pertuzumab.
Clinical Pharmacology for Perjeta
Mechanism Of Action
Pertuzumab targets the extracellular dimerization domain (Subdomain II) of the human epidermal growth factor receptor 2 protein (HER2) and, thereby, blocks ligand-dependent heterodimerization of HER2 with other HER family members, including EGFR, HER3, and HER4. As a result, pertuzumab inhibits ligand-initiated intracellular signaling through two major signal pathways, mitogen-activated protein (MAP) kinase, and phosphoinositide 3-kinase (PI3K). Inhibition of these signaling pathways can result in cell growth arrest and apoptosis, respectively. In addition, pertuzumab mediates antibody-dependent cell-mediated cytotoxicity (ADCC).
While pertuzumab alone inhibited the proliferation of human tumor cells, the combination of pertuzumab and trastuzumab augmented anti-tumor activity in HER2-overexpressing xenograft models.
Pharmacokinetics
Pertuzumab demonstrated linear pharmacokinetics at a dose range of 2 – 25 mg/kg. Based on a population PK analysis that included 481 patients, the median clearance (CL) of pertuzumab was 0.24 L/day and the median half-life was 18 days. With an initial dose of 840 mg followed by a maintenance dose of 420 mg every three weeks thereafter, the steady-state concentration of pertuzumab was reached after the first maintenance dose.
The population PK analysis suggested no PK differences based on age, gender, ethnicity (Japanese vs. non-Japanese), or disease status (neoadjuvant or adjuvant vs. metastatic setting). Baseline serum albumin level and lean body weight as covariates only exerted a minor influence on PK parameters. Therefore, no dose adjustments based on body weight or baseline albumin level are needed.
No dedicated renal impairment trial for PERJETA has been conducted. Based on the results of the population pharmacokinetic analysis, pertuzumab exposure in patients with mild (CLcr 60 to 90 mL/min, n=200) and moderate renal impairment (CLcr 30 to 60 mL/min, n=71) were similar to those in patients with normal renal function (CLcr greater than 90 mL/min, n=200). No relationship between CLcr and pertuzumab exposure was observed over the range of observed CLcr (27 to 244 mL/min).
Cardiac Electrophysiology
The effect of pertuzumab with an initial dose of 840 mg followed by a maintenance dose of 420 mg every three weeks on QTc interval was evaluated in a subgroup of 20 patients with HER2-positive breast cancer in CLEOPATRA. No large changes in the mean QT interval (i.e., greater than 20 ms) from placebo based on Fridericia correction method were detected in the trial. A small increase in the mean QTc interval (i.e., less than 10 ms) cannot be excluded because of the limitations of the trial design.
Clinical Studies
Metastatic Breast Cancer
CLEOPATRA (NCT00567190) was a multicenter, double-blind, placebo-controlled trial of 808 patients with HER2-positive metastatic breast cancer. HER2 overexpression was defined as a score of 3+ IHC or FISH amplification ratio of 2.0 or greater as determined by a central laboratory. Patients were randomly allocated 1:1 to receive placebo plus trastuzumab and docetaxel or PERJETA plus trastuzumab and docetaxel. Randomization was stratified by prior treatment (prior or no prior adjuvant/neoadjuvant anti-HER2 therapy or chemotherapy) and geographic region (Europe, North America, South America, and Asia). Patients with prior adjuvant or neoadjuvant therapy were required to have a disease-free interval of greater than 12 months before trial enrollment.
PERJETA was given intravenously at an initial dose of 840 mg, followed by 420 mg every 3 weeks thereafter. Trastuzumab was given intravenously at an initial dose of 8 mg/kg, followed by 6 mg/kg every 3 weeks thereafter. Patients were treated with PERJETA and trastuzumab until progression of disease, withdrawal of consent, or unacceptable toxicity. Docetaxel was given as an initial dose of 75 mg/m² by intravenous infusion every 3 weeks for at least 6 cycles.  The docetaxel dose could be escalated to 100 mg/m² at the investigator’s discretion if the initial dose was well tolerated. At the time of the primary analysis, the mean number of cycles of study treatment administered was 16.2 in the placebo-treated group and 19.9 in the PERJETA-treated group.
The primary endpoint of CLEOPATRA was progression-free survival (PFS) as assessed by an independent review facility (IRF). PFS was defined as the time from the date of randomization to the date of disease progression or death (from any cause) if the death occurred within 18 weeks of the last tumor assessment. Additional endpoints included overall survival (OS), PFS (investigator-assessed), objective response rate (ORR), and duration of response.
Patient demographic and baseline characteristics were balanced between the treatment arms. The median age was 54 (range 22 to 89 years), 59% were White, 32% were Asian, and 4% were Black. All were women with the exception of 2 patients. Seventeen percent of patients were enrolled in North America, 14% in South America, 38% in Europe, and 31% in Asia. Tumor prognostic characteristics, including hormone receptor status (positive 48%, negative 50%), presence of visceral disease (78%) and non-visceral disease only (22%) were similar in the study arms. Approximately half of the patients received prior adjuvant or neoadjuvant anti-HER2 therapy or chemotherapy (placebo 47%, PERJETA 46%). Among patients with hormone receptor positive tumors, 45% received prior adjuvant hormonal therapy and 11% received hormonal therapy for metastatic disease. Eleven percent of patients received prior adjuvant or neoadjuvant trastuzumab.
CLEOPATRA demonstrated a statistically significant improvement in IRF-assessed PFS in the PERJETA-treated group compared with the placebo-treated group [hazard ratio (HR)=0.62 (95% CI: 0.51, 0.75), p < 0.0001] and an increase in median PFS of 6.1 months (median PFS of 18.5 months in the PERJETA-treated group vs. 12.4 months in the placebo-treated group) (see Figure 1). The results for investigator-assessed PFS were comparable to those observed for IRF assessed PFS.
Consistent results were observed across several patient subgroups including age (< 65 or ≥ 65 years), race, geographic region, prior adjuvant/neoadjuvant anti-HER2 therapy or chemotherapy (yes or no), and prior adjuvant/neoadjuvant trastuzumab (yes or no). In the subgroup of patients with hormone receptor-negative disease (n=408), the hazard ratio was 0.55 (95% CI: 0.42, 0.72). In the subgroup of patients with hormone receptor-positive disease (n=388), the hazard ratio was 0.72 (95% CI: 0.55, 0.95). In the subgroup of patients with disease limited to non-visceral metastasis (n=178), the hazard ratio was 0.96 (95% CI: 0.61, 1.52). At the time of the final PFS analysis, 165 patients had died, and more deaths had occurred in the placebo-treated group (23.6%) compared with the PERJETA-treated group (17.2%); OS was not mature and interim OS analysis results did not meet the pre-specified stopping boundary for statistical significance. The final analysis of OS (Table 8, Figure 2) was performed when 389 patients had died (221 in the placebo-treated group and 168 in the PERJETA-treated group). A statistically significant OS improvement in favor of the PERJETA-treated group was demonstrated [HR=0.68 (95% CI; 0.56, 0.84), p=0.0002] with an increase in median OS of 15.7 months (median OS of 56.5 months in the PERJETA-treated group vs. 40.8 months in the placebo-treated group). OS results in patient subgroups were consistent with those observed for IRF-assessed PFS with the exception of the subgroup of patients with disease limited to nonvisceral metastasis [HR=1.11 (95% CI: 0.66, 1.85)].
Table 8 : Summary of Efficacy from CLEOPATRA
| Parameter | PERJETA + trastuzumab + docetaxel n=402 |
Placebo + trastuzumab + docetaxel n=406 |
HR (95% CI) | p-value |
| Progression-Free Survival (independent review) | ||||
| No. of patients with an event Median months | 191 (47.5%) 18.5 | 242 (59.6%) 12.4 | 0.62 (0.51, 0.75) | < 0.0001 |
| Overall Survival* (final analysis) | ||||
| No. of patients who died Median months | 168 (41.8%) 56.5 | 221 (54.4%) 40.8 | 0.68 (0.56, 0.84) | 0.0002 |
| Objective Response Rate (ORR, independent review) No. of patients analyzed | 343 | 336 | ||
| Objective response (CR + PR) | 275 (80.2%) | 233 (69.3%) | ||
| Complete response (CR) | 19 (5.5%) | 14 (4.2%) | ||
| Partial Response (PR) | 256 (74.6%) | 219 (65.2%) | ||
| Median Duration of Response (months) | 20.2 | 12.5 | ||
| Difference in ORR 95% CI | 10.8% (4.2%, 17.5%) | 0.0011 | ||
| * Final analysis of overall survival, cutoff date Feb 2014 CI=Confidence Interval |
||||
Figure 1 : Kaplan-Meier Curve of IRF-Assessed Progression-Free Survival for CLEOPATRA
 |
Figure 2 : Kaplan-Meier Curve of Overall Survival for CLEOPATRA (Final Analysis)
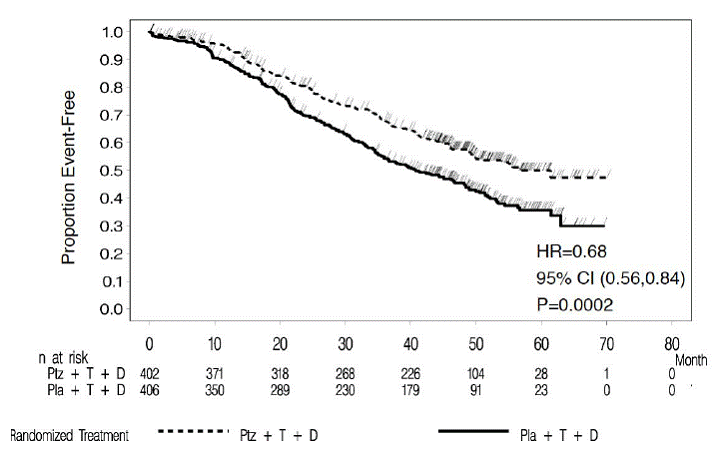 |
Neoadjuvant Treatment Of Breast Cancer
NeoSphere
NeoSphere (NCT00545688) was a multicenter, randomized trial conducted in 417 patients with operable, locally advanced, or inflammatory HER2-positive breast cancer (T2-4d) who were scheduled for neoadjuvant therapy. HER2 overexpression was defined as a score of 3+ IHC or FISH amplification ratio of 2.0 or greater as determined by a central laboratory. Patients were randomly allocated to receive 1 of 4 neoadjuvant regimens prior to surgery as follows: trastuzumab plus docetaxel, PERJETA plus trastuzumab and docetaxel, PERJETA plus trastuzumab, or PERJETA plus docetaxel. Randomization was stratified by breast cancer type (operable, locally advanced, or inflammatory) and estrogen receptor (ER) or progesterone receptor (PgR) positivity.
PERJETA was given intravenously at an initial dose of 840 mg, followed by 420 mg every 3 weeks for 4 cycles. Trastuzumab was given intravenously at an initial dose of 8 mg/kg, followed by 6 mg/kg every 3 weeks for 4 cycles. Docetaxel was given as an initial dose of 75 mg/m² by intravenous infusion every 3 weeks for 4 cycles. The docetaxel dose could be escalated to 100 mg/m² at the investigator’s discretion if the initial dose was well tolerated. Following surgery all patients received 3 cycles of 5-fluorouracil (600 mg/m²), epirubicin (90 mg/m²), and cyclophosphamide (600 mg/m²) (FEC) given intravenously every 3 weeks and trastuzumab administered intravenously every 3 weeks to complete 1 year of therapy. After surgery, patients in the PERJETA plus trastuzumab arm received docetaxel every 3 weeks for 4 cycles prior to FEC.
The primary endpoint of the study was pathological complete response (pCR) rate in the breast (ypT0/is). The FDA-preferred definition of pCR is the absence of invasive cancer in the breast and lymph nodes (ypT0/is ypN0).
Demographics were well balanced (median age was 49 – 50 years old, the majority were Caucasian (71%) and all were female. Overall, 7% of patients had inflammatory cancer, 32% had locally advanced cancer, and 61% had operable cancer. Approximately half the patients in each treatment group had hormone receptor-positive disease (defined as ER-positive and/or PgRpositive).
The efficacy results are summarized in Table 9. Statistically significant improvements in Pcr rates by both the study and FDA-preferred definitions were observed in patients receiving PERJETA plus trastuzumab and docetaxel compared to patients receiving trastuzumab plus docetaxel. The pCR rates and magnitude of improvement with PERJETA were lower in the subgroup of patients with hormone receptor-positive tumors compared to patients with hormone receptor-negative tumors.
Table 9 : Summary of Efficacy from NeoSphere
| Endpoint/Study Population | H+T | Ptz+H+T | Ptz+H | Ptz+T |
| Overall ITT | N=107 | N=107 | N=107 | N=96 |
| pCR1, n | 23 | 42 | 12 | 17 |
| (%) | (21.5%) | (39.3%) | (11.2%) | (17.7%) |
| [95% CI]2 | [14.1, 30.5] | [30.0, 49.2] | [5.9, 18.8] | [10.7, 26.8] |
| p-value (with Simes correction for CMH test)3 | 0.0063 (vs. H+T) |
0.0223 (vs. H+T) |
0.0018 (vs. Ptz+H+T) |
|
| Hormone receptor-positive subgroup | N=50 | N=50 | N=514 | N=46 |
| pCR1, n | 6 | 11 | 1 | 4 |
| (%) | (12.0%) | (22.0%) | (2.0%) | (8.7%) |
| [95% CI]2 | [4.5, 24.3] | [11.5, 36.0] | [0.1, 10.5] | [2.4, 20.8] |
| Hormone receptor-negative subgroup | N=57 | N=57 | N=554 | N=50 |
| pCR1, n | 17 | 31 | 11 | 13 |
| (%) | (29.8%) | (54.4%) | (20.0%) | (26.0%) |
| [95% CI]2 | [18.4, 43.4] | [40.7, 67.6] | [10.4, 33.0] | [14.6, 40.3] |
| T=docetaxel, Ptz=PERJETA, H=trastuzumab CI=Confidence Interval 1 ypT0/is ypN0 (absence of invasive cancer in the breast and lymph nodes) 2 95% CI for one sample binomial using Pearson-Clopper method. 3 p-value from Cochran-Mantel-Haenszel (CMH) test, with Simes multiplicity adjustment 4 One patient had unknown hormone receptor status. The patient did not achieve a pCR. |
||||
TRYPHAENA
An additional neoadjuvant study (TRYPHAENA, NCT00976989) was conducted in 225 patients with HER2-positive locally advanced, operable, or inflammatory (T2-4d) breast cancer designed primarily to assess cardiac safety in which all arms included PERJETA. HER2 overexpression was defined as a score of 3+ IHC or FISH amplification ratio of 2.0 or greater as determined by a central laboratory.
Patients were randomly allocated to receive 1 of 3 neoadjuvant regimens prior to surgery as follows: 3 cycles of FEC followed by 3 cycles of docetaxel all in combination with PERJETA and trastuzumab, 3 cycles of FEC alone followed by 3 cycles of docetaxel and trastuzumab in combination with PERJETA, or 6 cycles of docetaxel, carboplatin, and trastuzumab (TCH) in combination with PERJETA. Randomization was stratified by breast cancer type (operable, locally advanced, or inflammatory) and ER and/or PgR positivity.
PERJETA was given by intravenous infusion at an initial dose of 840 mg, followed by 420 mg every 3 weeks. Trastuzumab was given by intravenous infusion at an initial dose of 8 mg/kg, followed by 6 mg/kg every 3 weeks. 5-Fluorouracil (500 mg/m²), epirubicin (100 mg/m²), and cyclophosphamide (600 mg/m²) were given intravenously every 3 weeks for 3 cycles. In the PERJETA plus trastuzumab, docetaxel, and FEC arms, docetaxel was given as an initial dose of 75 mg/m² by intravenous infusion every 3 weeks for 3 cycles with the option to escalate to 100 mg/m² at the investigator’s discretion if the initial dose was well tolerated. However, in the PERJETA plus TCH arm, docetaxel was given intravenously at 75 mg/m² (no escalation was permitted) and carboplatin (AUC 6) was given intravenously every 3 weeks for 6 cycles. Following surgery all patients received trastuzumab to complete 1 year of therapy, which was administered intravenously every 3 weeks.
Demographics were well balanced (median age was 49-50 years old, the majority were Caucasian [76%]) and all were female. Overall 6% of patients had inflammatory cancer, 25% had locally advanced cancer and 69% had operable cancer, with approximately half the patients in each treatment group having ER-positive and/or PgR-positive disease.
The pCR (ypT0/is ypN0) rates were 56.2% (95% CI: 44.1%, 67.8%), 54.7% (95% CI: 42.7%, 66.2%), and 63.6% (95% CI: 51.9%, 74.3%) for patients treated with PERJETA plus trastuzumab and FEC followed by PERJETA plus trastuzumab and docetaxel, PERJETA plus trastuzumab and docetaxel following FEC, or PERJETA plus TCH, respectively. The pCR rates were lower in the subgroups of patients with hormone receptor-positive tumors: 41.0% (95% CI: 25.6%, 57.9%), 45.7% (95% CI: 28.8%, 63.4%), and 47.5% (95% CI: 31.5%, 63.9%) than with hormone receptor-negative tumors: 73.5% (95% CI: 55.6%, 87.1%), 62.5% (95% CI: 45.8%, 77.3%), and 81.1% (95% CI: 64.8%, 92.0%), respectively.
BERENICE
A two-arm non-randomized study (BERENICE, NCT02132949) was conducted in 401 patients with HER2-positive locally advanced, inflammatory, or early-stage HER2-positive breast cancer. HER2 overexpression was defined as a score of 3+ IHC or ISH amplification ratio of 2.0 or greater as determined by a central laboratory.
Patients received 1 of 2 neoadjuvant regimens prior to surgery as follows: 4 cycles of dose dense doxorubicin and cyclophosphamide (ddAC) followed by 4 cycles of PERJETA in combination with trastuzumab and weekly paclitaxel for 12 weeks or 4 cycles of 5-fluorouracil, epirubicin and cyclophosphamide (FEC) followed by 4 cycles of PERJETA in combination with trastuzumab and docetaxel. The choice of neoadjuvant treatment regimen was made by the Investigator on a site-specific basis. Dosing for the regimens was as follows:
- PERJETA was given by intravenous infusion at an initial dose of 840 mg, followed by 420 mg every 3 weeks. Trastuzumab was given by intravenous infusion at an initial dose of 8 mg/kg, followed by 6 mg/kg every 3 weeks.
- In the ddAC cohort, (doxorubicin 60 mg/m² and cyclophosphamide 600 mg/m²) were given intravenously every 2 weeks (ddAC) for 4 cycles with G-CSF (granulocyte colony stimulating factor) support at investigator discretion, followed by paclitaxel 80 mg/m² given intravenously weekly for 12 weeks, with PERJETA and trastuzumab every 3 weeks from the start of paclitaxel for 4 cycles.
- In the FEC cohort, 5-Fluorouracil (5-FU) (500 mg/m²), epirubicin (100 mg/m²), and cyclophosphamide (600 mg/m²) were given intravenously every 3 weeks for 4 cycles, followed by docetaxel given as an initial dose of 75 mg/m² by intravenous infusion every 3 weeks for 4 cycles with PERJETA and trastuzumab, and with the option to escalate to 100 mg/m² at the investigator’s discretion if the initial dose was well tolerated.
Following surgery, all patients received PERJETA and trastuzumab administered intravenously every 3 weeks to complete 1 year of therapy.
The median age of the overall study population was 49 years old (range 21-78), 12% of patients were 65 or older, 83% were Caucasian, and all but one patient was female. Overall 3% of patients had inflammatory cancer, 23% had locally advanced cancer (Stage 3A or greater), 5% were not classified per TNM staging, with approximately two thirds of the patients in each treatment group having ER-positive and/or PgR-positive disease. All patients had an ECOG performance status of 0 or 1.
The pCR (ypT0/is ypN0) rates were 61.8% (95% CI: 54.7, 68.6) and 60.7% (95% CI: 53.6, 67.5) for patients treated with ddAC followed by PERJETA plus trastuzumab and paclitaxel, or FEC followed by PERJETA plus trastuzumab and docetaxel, respectively. The pCR rates were lower in the subgroups of patients with hormone receptor-positive tumors: 51.6% (95% CI: 42.6, 60.5%) and 57.3% (95% CI: 48.1, 66.1%) than with hormone receptor-negative tumors: 81.5% (95% CI: 70.0, 90.1%) and 68.0% (95% CI: 56.2, 78.3%), respectively.
Adjuvant Treatment Of Breast Cancer
APHINITY (NCT01358877) was a multicenter, randomized, double-blind, placebo-controlled study conducted in 4804 patients with HER2-positive early breast cancer who had their primary tumor excised prior to randomization. Patients were then randomized to receive PERJETA or placebo, in combination with adjuvant trastuzumab and chemotherapy. Randomization was stratified by the following factors: region, nodal status, protocol version, central hormone receptor status, and adjuvant chemotherapy regimen.
Investigators selected one of the following anthracycline-based or non-anthracycline-based chemotherapy regimens for individual patients:
- 3 or 4 cycles of FEC (5-FU 500-600 mg/m², epirubicin 90-120 mg/m², cyclophosphamide 500-600 mg/m²) or FAC (5-FU 500-600 mg/m², doxorubicin 50 mg/m², cyclophosphamide 500-600 mg/m²), followed by 3 or 4 cycles of docetaxel (75 mg/m² which could be escalated to 100 mg/m² every 3 weeks) or 12 cycles of weekly paclitaxel (80 mg/m²).
- 4 cycles of AC (doxorubicin 60 mg/m² and cyclophosphamide 500-600 mg/m²) or EC (epirubicin 90-120 mg/m² and cyclophosphamide 500-600 mg/m²) either every 3 weeks or every 2 weeks with GCSF support, followed by docetaxel (100 mg/m² for 3 cycles or 75 mg/m² for first cycle and 100 mg/m² for subsequent three cycles, or 75 mg/m² for four cycles) or 12 cycles of weekly paclitaxel (80 mg/m²).
- 6 cycles of docetaxel (75 mg/m²) in combination with carboplatin (AUC 6) PERJETA and trastuzumab were administered intravenously every 3 weeks starting on Day 1 of the first taxane-containing cycle, for a total of 52 weeks (up to 18 cycles) or until recurrence, withdrawal of consent, or unmanageable toxicity.
After completion of chemotherapy, patients received radiotherapy and/or hormone therapy as per investigator’s discretion.
The major efficacy outcome of the study was invasive disease-free survival (IDFS), defined as the time from randomization to first occurrence of ipsilateral local or regional invasive breast cancer recurrence, distant recurrence, contralateral invasive breast cancer, or death from any cause. Additional efficacy endpoints were IDFS including second primary non-breast cancer, disease-free survival (DFS), and overall survival (OS).
Demographics were generally balanced between the two treatment arms. The median age was 51 years (range 18-86), 13% of patients were 65 or older, and over 99% of patients were female. Sixty-three percent of patients had node-positive disease, 64% had hormone receptor-positive disease, and 71% were Caucasian. All patients had an ECOG performance status of 0 or 1. Â Seventy-eight percent received an anthracycline containing regimen.
PERJETA-treated patients and placebo-treated patients both received a median number of 18 cycles of anti-HER2 therapy. After a median follow-up of 45.4 months, a statistically significant improvement in IDFS was demonstrated in patients randomized to receive PERJETA compared with patients randomized to receive placebo. The efficacy results from APHINITY are summarized in Tables 10 and 11 and in Figure 3.
Table 10 : Efficacy Results from APHINITY
| PERJETA + trastuzumab + chemotherapy N=2400 |
Placebo + trastuzumab + chemotherapy N=2404 |
|
| Invasive Disease Free Survival (IDFS) | ||
| Number (%) of patients with event | 171 (7.1%) | 210 (8.7%) |
| HR [95% CI] 1 | 0.82 [0.67, 1.00] | |
| p-value (Log-Rank test, stratified1) | 0.047 | |
| 3 year event-free rate2, % [95% CI] | 94.1 [93.1, 95.0] | 93.2 [92.2, 94.3] |
| IDFS including second primary non-breast cancer | ||
| Number (%) of patients with event | 189 (7.9%) | 230 (9.6%) |
| HR [95% CI] 1 | 0.83 [0.68, 1.00] | |
| 3 year event-free rate2, % [95% CI] | 93.5 [92.5, 94.5] | 92.5 [91.4, 93.6] |
| Disease Free Survival (DFS) | ||
| Number (%) of patients with event | 192 (8.0%) | 236 (9.8%) |
| HR [95% CI] 1 | 0.82 [0.68, 0.99] | |
| 3 year event-free rate2, % [95% CI] | 93.4 [92.4, 94.4] | 92.3 [91.2, 93.4] |
| Overall Survival (OS)3 | ||
| Number (%) of patients with event | 80 (3.3%) | 89 (3.7%) |
| HR [95% CI] 1 | 0.89 [0.66, 1.21] | |
| 3 year event-free rate2, % [95% CI] | 97.7 [97.0, 98.3] | 97.7 [97.1, 98.3] |
| HR=Hazard Ratio, CI=Confidence Interval 1 All analyses stratified by nodal status, protocol version, central hormone receptor status, and adjuvant chemotherapy regimen. Stratification factors are defined according to the randomization data for IDFS. 2 3-year event-free rate derived from Kaplan-Meier estimates 3 Data from first interim analysis |
||
Figure 3 : Kaplan-Meier Curve of Invasive Disease Free Survival from APHINITY (ITT Population)
 |
Table 11 : Efficacy Results by Baseline Disease Characteristics and Adjuvant Chemotherapy from APHINITY1
| Population | Number of events/Total N (%) | IDFS at 3 year (%, 95% CI) | Unstratified HR (95% CI) | ||
| PERJETA + trastuzumab + chemotherapy | Placebo + trastuzumab + chemotherapy | PERJETA + trastuzumab + chemotherapy | Placebo + trastuzumab + chemotherapy | ||
| Hormone Receptor Status | |||||
| Negative | 71/864 (8.2%) |
91/858 (10.6%) |
92.8 (90.8, 94.3) |
91.2 (89.0, 92.9) |
0.76 (0.56, 1.04) |
| Positive | 100/1536 (6.5%) |
119/1546 (7.7%) |
94.8 (93.5, 95.8) |
94.4 (93.1, 95.4) |
0.86 (0.66, 1.13) |
| Nodal Status | |||||
| Negative | 32/897 (3.6%) |
29/902 (3.2%) |
97.5 (96.3, 98.4) |
98.4 (97.3, 99.0) |
1.13 (0.68, 1.86) |
| Positive | 139/1503 (9.2%) |
181/1502 (12.1%) |
92.0 (90.5, 93.3) |
90.2 (88.5, 91.6) |
0.77 (0.62, 0.96) |
| Adjuvant Chemotherapy Regimen | |||||
| Anthracycline | 139/1865 (7.4%) |
171/1877 (9.1%) |
93.8 (92.6, 94.8) |
93.0 (91.8, 94.1) |
0.82 (0.66, 1.03) |
| Non-Anthracycline | 32/535 (6.0%) |
39/527 (7.4%) |
94.9 (92.6, 96.6) |
94.0 (91.5, 95.8) |
0.82 (0.51, 1.31) |
| 1Exploratory analyses without adjusting multiple comparisons, therefore, results are considered descriptive. | |||||
Patient Information for Perjeta
Left Ventricular Dysfunction
Advise patients to contact a health care professional immediately for any of the following: new onset or worsening shortness of breath, cough, swelling of the ankles/legs, swelling of the face, palpitations, weight gain of more than 5 pounds in 24 hours, dizziness or loss of consciousness [see WARNINGS AND PRECAUTIONS].
Embryo-Fetal Toxicity
Advise pregnant women and females of reproductive potential that exposure to PERJETA in combination with trastuzumab during pregnancy or within 7 months prior to conception can result in fetal harm. Advise female patients to contact their healthcare provider with a known or suspected pregnancy [see Use In Specific Populations].
Advise women who are exposed to PERJETA in combination with trastuzumab during pregnancy or within 7 months prior to conception that there is a pregnancy pharmacovigilance program that monitors pregnancy outcomes. Encourage these patients to report their pregnancy to Genentech [see Use In Specific Populations].
Advise females of reproductive potential to use effective contraception during treatment and for 7 months following the last dose of PERJETA in combination with trastuzumab [see Use In Specific Populations].
From 
Report Problems to the Food and Drug Administration
You are encouraged to report negative side effects of prescription drugs to the FDA. Visit the FDA MedWatch website or call 1-800-FDA-1088.